
Co-processing of bio-oil from de-oiled Jatropha curcas seed cake with refinery gas–oil over sulfided CoMoP/Al2O3 catalyst†
Mukesh Kumar
Poddar
ab,
Aditya
Rai
a,
Mannar Ram
Maurya
b and
Anil Kumar
Sinha
*a
aCSIR-Indian Institute of Petroleum, Dehradun, India. E-mail: asinha@iip.res.in; Tel: +91-1352525842
bIndian Institute of Technology, Roorkee, India
First published on 23rd November 2016
Abstract
A sulfided cobalt–molybdenum–phosphorus/aluminium oxide (CoMoP/Al2O3) catalyst was studied in the hydroprocessing of bio-oil (BO) obtained from the pyrolysis of de-oiled Jatropha curcas seed cake. Hydroprocessing was carried out with different ratios of refinery gas oil (GO) and BO. The oxygen content in the products was reduced to trace amounts after hydroprocessing. A clear product obtained from the co-processing of BO with refinery GO contained 2–16% gasoline, 30–35% kerosene, 35–44% diesel, with 50–60% alkanes, 10–45% cycloalkanes, and 1–10% aromatics, with a negligible amount of char formed in the process. Hydroprocessing of 100% BO produced 30% kerosene and 30% diesel, together with 10% gasoline, with 15% of alkanes and 15% cycloalkanes, and 45% aromatics. A maximum amount of kerosene (41%) was obtained at 648 K and 75 bar from 100% BO, with a small amount of char (1.5%) deposited on the catalyst. In comparison, over sulfided CoMo/Al2O3 catalyst (without P promoter) only 31% of kerosene was produced, with 17% char, using similar reaction conditions.
1. Introduction
Liquid transportation fuels (hydrocarbons) from biomass are renewable alternatives to the current fossil derived fuels. Diverse technologies have been investigated for the conversion of biomass into liquid fuels, such as biodiesel from plant oils and bio-ethanol from carbohydrate rich resources.1 Because of the food versus fuel issues, these first generation biofuels were not encouraged and new options are being developed.1 Second generation biofuel platforms are thermochemical biomass conversion routes such as pyrolysis, gasification and hydrothermal liquefaction of agricultural wastes, forestry wastes and municipal wastes and so on.1 High abundance, availability and low procurement cost are the advantages for considering biomass as a source. There is great potential for successful deployment of technologies to produce a liquid biofuel from biomass with cost reductions.2Liquid product yields from the pyrolysis of de-oiled Jatropha curcas cake (non-edible) seeds is up to 60–70 wt% and the product (bio-oil; BO) consists of a wide range of functional groups containing oxygenate and heavy compounds with a wide range of molecular weights.3,4 Classes of compounds in BO are organic acids, aldehydes, ketones, phenolics and alcohols. The BO required upgrading because as such it was not suitable as a biofuel for high speed internal combustion engines, because of large amounts of water (up to 30 wt%) and corrosive organic acids (up to 10 wt%) present in it.5,6 It also has limited storage stability and undesirable physical properties.
Catalytic BO upgrading presently seems to be a techno-economic process towards production of fuel-like components. A major aim of upgrading BO is to convert the oxygen-rich, high molecular weight components into hydrocarbons that are similar to petroleum-derived fuels (drop-in fuels). Upgrading is done using chemical methods (such as cracking and so on), and physical methods (such as distillation and so on).6,7 Catalytic hydrotreatment is considered to be a promising upgrading technology to produce drop-in kind fuels from BO.8 This process involves treatment of BO with hydrogen (H2) in the presence of a heterogeneous catalyst. However, selection of stable and productive catalyst(s) towards refinery products with low coke formation is a great challenge. The primary aim is minimization of the oxygen content and reduction of the chain length by a process called hydroprocessing (catalytic high pressure hydrotreatment or hydrodeoxygenation accompanied by hydrocracking). Hydrodeoxygenation is a chemical conversion that takes place at high H2 partial pressures (as high as 75–300 bar) and high temperatures (523–723 K) to remove oxygen primarily in the form of water or carbon dioxide (CO2).9,10
Hydrodeoxygenation of BO gives hydrocarbons and oxygen containing organic compounds in addition to water and CO2, and the classes of reactions include decarboxylation, hydrogenation, hydrogenolysis, hydrocracking, and dehydration leading to gasoline, kerosene and diesel like products. Research activities on the hydrodeoxygenation of pyrolysis oil started in 1984 with the pioneering work of Elliott and Baker on commercial hydrodesulfurization (HDS) catalysts, i.e., sulfided nickel–molybdenum/aluminium oxide (NiMo/Al2O3) and cobalt–molybdenum (CoMo)/Al2O3.8 The primary issue interfering with long-term operation of these systems was the fouling of the catalyst bed by carbonaceous deposits. Deactivation of these catalysts as a function of time on stream is reported.11–13 Deactivation might be because of the blockage of catalyst pores and active sites, sintering of the active metals, poisoning of the catalyst, structural degradation of the support and active sites, coking, and metal deposition.12,14
This paper reports the production of transportation fuels (gasoline, kerosene and diesel) by processing a heavy, dark viscous liquid (BO) obtained from waste de-oiled J. curcas seeds made into a cake, or with refinery streams, by using the economically viable catalyst sulfided cobalt–molybdenum–phosphorus (CoMoP)/Al2O3.15 The advantage of this work is that transportation fuels can be produced by using a single catalyst instead of other expensive multi-catalyst processes such as hydrodeoxygenation with noble metals followed by cracking. It is demonstrated that the optimization of process conditions in this work has resulted in suppression of carbonaceous deposits by minimizing the polynuclear aromatics formation which are the precursors for carbonaceous deposits.
2. Experimental details
2.1 Materials
Cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O), ammonium molybdate tetrahydrate [(NH4)6Mo7O24·4H2O] and orthophosphoric acid (85% of aq. H3PO4) were obtained from Sigma-Aldrich. The γ-alumina was obtained from Sasol. High-performance liquid chromatography (HPLC) grade water was used as a solvent for synthesis. BO was produced in a tubular reactor (using Swagelok SS 314 tubing) [shown in Fig. S1; ESI†]. Gas–oil (GO) was obtained from Mathura Refinery, India.2.2 Catalyst preparation
The catalyst was prepared using an incipient wet impregnation method. The calculated amount of the Mo metal precursor (NH4)6Mo7O24·4H2O (1.89 g) in liquid ammonia was added dropwise, with continuous stirring to 7.9 g of dried γ-alumina to obtain the required amount of Mo (16 wt%) loading on the support. Then the calculated amount of Co metal precursor (Co(NO3)2·6H2O, 1.55 g) solution in water was added dropwise with continuous stirring to obtain the required amount of Co (4 wt%). The solution was dried at 393 K for 4 h. After drying, the calculated amount of 85% aq. H3PO4 (0.16 g) was added dropwise with continuous stirring to obtain the required amount of P (1 wt%). The sample was dried at 393 K and calcined at 823 K for 6 h (heating rate, 1 K min−1).2.3 Characterization methods
The surface area of the catalyst was measured using the Brunauer–Emmett–Teller (BET) method with a nitrogen (N2) adsorption isotherm at 77 K using BELSORP-max Microtrac (Japan) apparatus. Before analysis the catalyst sample was degassed at 523 K under vacuum. The pore size was calculated from the desorption isotherm using the Barrett, Joyner and Hallender (BJH) method.Samples for transmission electron microscopy (TEM) were prepared by deposition of the catalyst, suspended in isopropanol, on a copper grid by. TEM images were recorded using a Tecnai G2 (FEI) operating at 200 kV. Deposited metals (Co 4%, Mo 16% and P 1%) were determined using an inductively coupled plasma atomic emission spectroscopy method on a PS 3000 UV (DRE) (Teledyne Leeman Labs, Inc., USA).
2.4 Reaction procedure
2.5 Analysis
As in normal petroleum refinery practice, the product distributions were quantified on the basis of hydrocarbon size (carbon numbers): gasoline (![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) C9 hydrocarbons), kerosene (C9–C14 hydrocarbons), diesel (C15–C18 hydrocarbons) and heavy oil (
C9 hydrocarbons), kerosene (C9–C14 hydrocarbons), diesel (C15–C18 hydrocarbons) and heavy oil (![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C18 hydrocarbons). Errors up to ±4% were observed, and are included in the final results.
C18 hydrocarbons). Errors up to ±4% were observed, and are included in the final results.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :495:5 was used as solvent for the titration. For the titration of each sample, a blank titration of the solvent was also done, to obtain the relative TAN value of hydroprocessed BO with respect to the solvent. For each analysis 125 ml of solvent was taken. The weights of samples to be analyzed were 0.25–1.85 g and the repeatability of the weighing balance was ±0.00025 g. The TAN was determined three times for each sample, and the variation in TAN was ±1 to 2 mgKOH g−1.
:495:5 was used as solvent for the titration. For the titration of each sample, a blank titration of the solvent was also done, to obtain the relative TAN value of hydroprocessed BO with respect to the solvent. For each analysis 125 ml of solvent was taken. The weights of samples to be analyzed were 0.25–1.85 g and the repeatability of the weighing balance was ±0.00025 g. The TAN was determined three times for each sample, and the variation in TAN was ±1 to 2 mgKOH g−1.
3. Results and discussion
3.1 Catalyst characterization
The surface area (BET) of the catalyst support was 298 m2 g−1 with a pore volume of 0.5 ml g−1 and a mean pore size (BJH) of 7 nm. A TEM image of the sulfided CoMo/Al2O3 catalyst is shown in Fig. 1. The well dispersed, straight and curved CoMo–S nanoslabs of width 1–2 nm and length 20–60 nm can be seen in the TEM micrograph shown in Fig. 1. If the interlayer spacing is taken to be 0.65 nm, the CoMo–S nanoslabs consist of about two to three layers.17 For the CoMoS/Al2O3 catalyst, the typical CoMoS particle size is 5–8 nm long.17,18 This clearly indicates that the P used during impregnation has resulted in better active metal dispersion with smaller nanoslab sizes. | ||
| Fig. 1 TEM micrograph of sulfided CoMoP/Al2O3 catalyst. | ||
3.2 Catalytic studies
Elemental analysis (CHN) of the feeds and hydroprocessed products are listed in Table 1. The detailed composition of BO has been reported in the Table S1 (ESI†).16 De-oiled Jatropha seed cake contains 3–7% of triglyceride remnants after expeller extraction. The carbon content in the product was increased slightly from 78% to 80% and the H2 content increased from 12% to 13% when the reaction temperature was increased from 573 K to 673 K for the feed with 25% BO in GO. The nitrogen content was reduced from 7% to 3% for 100% BO hydroprocessing and it was reduced further to ∼1% for 25% and 50% BO in GO. The remaining 10% and 20% composition in GO and BO, were predominantly O and some S, respectively. The higher O content in GO could be because of oxidation, dissolved oxygen and moisture from prolonged storage under ambient conditions. Sulfur reduction from the feed (2300–2500 ppm S) was 50–80% (down to 400–1000 ppm S) during this co-processing study. In comparison, S removal was 80–90% for catalytic upgraded pristine GO. During the co-processing of oxygenated feeds with GO, deoxygenation reactions do not adversely affect the hydrodesulfurization reactions, as also reported by Rana et al.17 and Tiwari et al.19 The oxygen content in the O was 19% which reduced to trace amounts after hydroprocessing of all the feeds.| Feed composition | Conditions | Elemental composition | ||||
|---|---|---|---|---|---|---|
| BO (wt%) | GO (wt%) | Temp. (K) | Pressure (bar) | C (wt%) | H (wt%) | N (wt%) |
| Reactants | ||||||
| 100 | 0 | — | — | 64.5 | 8.8 | 6.7 |
| 0 | 100 | — | — | 76.6 | 13.3 | 0.04 |
|
||||||
| Products | ||||||
| 25 | 75 | 573 | 50 | 78.2 | 11.8 | 0.7 |
| 25 | 75 | 623 | 50 | 81.6 | 12.3 | 0.9 |
| 25 | 75 | 648 | 50 | 80.0 | 12.2 | 0.8 |
| 25 | 75 | 673 | 50 | 81.4 | 13.1 | 0.7 |
| 50 | 50 | 648 | 50 | 81.6 | 12.0 | 0.9 |
| 100 | 0 | 648 | 50 | 65.6 | 7.9 | 3.4 |
The product (liquid, solid and gases) distributions (yield%) of the hydroprocessing reactions of the feeds with 25%, 50% and 100% of BO mixed with refinery GO at various temperatures (573–673 K) and feed compositions (25%, 50% and 100% of BO in GO), over CoMoP/Al2O3, are given in Fig. 2 and 3, respectively. For the liquid product yields for 25% BO in GO (Fig. 2) at various temperatures and H2 pressure of 50 bar, a maximum liquid yield (63.4%) was obtained at 673 K, which was reduced to 18.2% at a lower temperature of 573 K. However, little change in the yields of gases with temperature was observed (maximum 23.5% at 673 K and 21.2% at 573 K). Char yield increased monotonously from 0% to 12% as temperature increased from 573 K to 673 K. The liquid yield for pure GO was 85–95%. The unprocessable BO content was very high at a lower reaction temperature of 573 K (∼60%) which reduced drastically (2–5%) at higher reaction temperatures (648–673 K). These results indicate that at an intermediate reaction temperature, the undesirable char formation is suppressed but a nearly complete BO conversion is obtained.
 | ||
| Fig. 2 Yield (%) of the products after reaction of 25% of BO in GO at various temperatures. | ||
 | ||
| Fig. 3 Yield (%) of products after reaction of feeds with 25%, 50% and 100% of BO in GO at 648 K. | ||
Fig. 3 shows the product distributions (char, liquid and gases) for the reaction of feeds of various compositions (25%, 50% and 100% BO in GO) at a temperature of 648 K. A maximum liquid product yield (63.4%) was obtained with 100% BO feed which also gave the maximum char yield (12.2%), whereas the maximum gas yield (40%) were observed for feed with 50% BO in GO. The hydroprocessed product, a transparent liquid, obtained as a phase separate from water, was composed of light and heavy fractions. The product distributions for 25% BO in GO at various temperatures and a of H2 pressure of 50 bar are shown in Fig. 4. The diesel yield increased with increase of reaction temperature from 573 K to 673 K because of the increasing severity of the reaction which favors cracking of BO components. At a reaction temperature of 673 K, the maximum gasoline (C9) (16%) and maximum diesel (C15–C18) (45%) were produced, whereas heavy oil (C18) yield was at its minimum (9%), because of severe cracking conditions.
 | ||
| Fig. 4 Distribution of hydrocarbons [○ gasoline (C9), △ kerosene (C9–C14), ▽ diesel (C15–C18), and ◊ heavy residue (C18)] in the products from feed containing 25% BO in GO at various temperatures and H2 pressure of 50 bar. | ||
As the severity of the reaction increased with increase in temperature the diesel yield increased monotonously (because deoxygenation reactions are favored). The longer chain components (C18) underwent cracking with increasing temperature and thus, their yield decreased monotonously from 33% at 573 K to 9% at 673 K.
Gasoline (C9) product yield showed little increase with increasing temperature from 573 K to 648 K. Beyond 648 K there was a rapid increase in gasoline yield from 2% (at 648 K) to 16% (at 673 K) because of increased reaction severity leading to more cracking. In contrast, the heavier products (C18 hydrocarbons) decreased little with increasing temperature (573–648 K), but there was sharp decrease in yield from 27% at 648 K to 9% at 673 K. These results indicated that at 673 K the heavier hydrocarbons (C18) crack selectively into the gasoline range (C9) hydrocarbons. Maximum aviation kerosene (C9–C14 hydrocarbons) yield (36%) was obtained at an intermediate temperature of 623 K. At higher temperatures (648–673 K) the kerosene yield decreased by about 5%. For 100% BO hydroprocessing, a high char content after reaction was a major problem (Fig. 3). Higher reaction pressures generally suppress char formation. It was observed that at higher pressures, not only more kerosene (41%) was obtained (at 648 K and 75 bar) from 100% BO but also that char formation was also suppressed (1.5%). These results indicate that it is necessary to operate at higher pressures for hydroprocessing pure BO to obtain desired products and to minimize undesired coking and in order to prolong the catalyst life. In comparison the CoMo/Al2O3 catalyst (without P promoter) showed only 31% kerosene and 17% char was obtained at similar reaction conditions. This indicates that P as promoter suppresses char formation and thus, improves the catalyst life. Catalyst life is dependent on char/coke deposition rate. Catalyst deactivation was mainly because of coke deposition over the catalyst. Char/coke formation over the P promoted catalyst was nearly 10 times lower than that over catalyst without P as promoter, which clearly indicates that the P promoted catalyst has better catalyst life. The catalyst modified by P works in two ways: there is an increase in active sites by enhanced metal dispersion and increase in Brönsted acidity, Mo interacts preferentially with the P–OH groups on P-added γ-alumina and also the other surface hydroxyls become more reactive with Mo in the presence of P.20,21 It was also observed that catalysts prepared by impregnating P, on CoMo/Al2O3 (co-impregnation) were more active than the P-free catalyst.22 Earlier work on BO hydrotreatment with nickel–molybdenum–sulfur (NiMoS)/Al2O3 has reported that at 648–723 K and 70 bar pressure, formation of polynuclear aromatics was rapid which prevented long-term operation under these conditions.19,23
Isomerisation is required to produce aviation kerosene with a desirable freezing point. Table 2 shows that the kerosene range product had high isomer selectivity with isomer and normal alkane ratios (i/n ratio) varying between 1 and 2. The highest iso-alkane yield (i/n = 2) was obtained at the lowest reaction temperature studied (573 K), for 25% BO in GO feed. Even though CoMoP/Al2O3 may not have required Brönsted acidity, the high TAN of the feed was expected to catalyze the isomerisation reactions during the hydrocracking of BO, as also reported earlier for the co-processing of oxygenated biomass derived oils with GO over a CoMo/Al2O3 catalyst.24
| Conditions | Distribution of alkanes in kerosene | |||
|---|---|---|---|---|
| Temp/K | % of BO (in GO) | n-Alkanes | i-Alkanes | i/n ratio |
| 573 | 25 | 10.95 | 19.47 | 2 |
| 623 | 25 | 18.53 | 16.97 | 1 |
| 623 | 50 | 11.62 | 11.5 | 1 |
| 648 | 25 | 13.16 | 18.44 | 1 |
| 648 | 50 | 12.53 | 16.94 | 1 |
| 648 | 100 | 13.98 | 16.31 | 1 |
The product yields for reaction of 25%, 50% and 100% BO in GO at 648 K are shown in Fig. 5. The maximum gasoline (C9) yield (13%) was obtained from the reaction of 100% BO at 648 K. The major components of BO are short-chain oxygenated compounds, and thus, as expected, the lighter components (C9) in the product increase with increasing BO% in the feed. There was little change in the product yields with variation in BO content in GO from 25% to 50%. Increasing BO in GO, from 50 to 100%, caused the diesel content to be reduced to 32% from 40%, whereas the kerosene yield showed little variation, and the gasoline (C9) yield increased from 3% to 13%, and the heavy oil components (C18) reduced slightly from 28% to 25%. These results indicated that pure BO (undiluted with GO) shows a completely different product pattern to the pattern obtained for BO mixed with GO. This may also be correlated with the large differences in the TAN of pure BO (52 mgKOH g−1) and the mixture of BO and GO (12 mgKOH g−1 for the 25% mixture and 23 mgKOH g−1 for the 50% mixture). The higher TAN for pure BO results in more acidity in the reaction mixture which would catalyze the cracking reactions, thus, higher cracked products (gasoline) yield and a lower diesel range hydrocarbons yields are observed. The higher TAN (acidity) feeds have been reported to show more cracking activity15,24 which was clearly correlated to the acidity of the feed, because the catalyst used had a very low acidity.
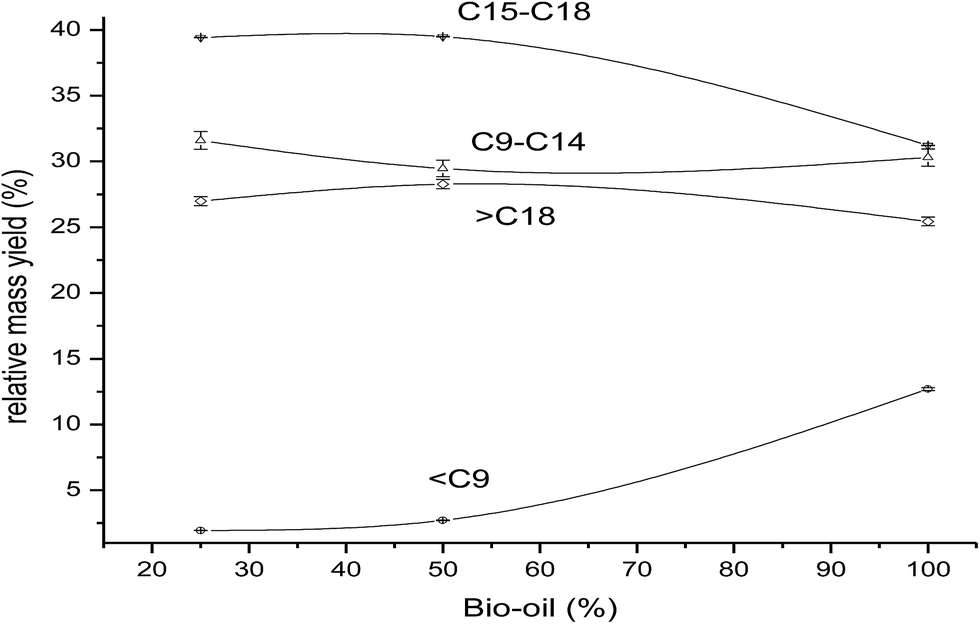 | ||
| Fig. 5 Distribution of hydrocarbons [○ gasoline (C9), △ kerosene (C9–C14), ▽ diesel (C15–C18), and ◊ heavy residue (C18)] in the products from feeds containing 25%, 50% and 100% of BO in GO (at 648 K, H2 pressure of 50 bar, 10 wt% of catalyst, 5 h). | ||
The distribution of alkanes, cycloalkanes, aromatics and polynuclear aromatics in hydroprocessed products obtained from GCxGC-MS analysis are shown in Fig. 6 and 7. GCxGC-MS analysis showed the absence of any observable oxygenated compounds indicating that there was complete oxygen removal from the BO components during hydroprocessing. Fig. 6 shows the distribution of hydrocarbon type for the feed with 25% of BO in GO at various temperatures and H2 pressure of 50 bar. The trend for the formation of cycloalkanes showed that the maximum cycloalkane yield (45%) was formed at a temperature of 673 K. The maximum alkane yield (63%) was formed at 648 K and alkane yield at 673 K was minimum (55%). The aromatics yield was zero at 673 K. There was little variation in the yield of various hydrocarbons at temperatures from 573 K to 648 K. Aromatics are thermodynamically controlled product and thus, their yield was minimum at a higher reaction temperature of 673 K. More cracking was favoured at a higher temperature (673 K) and thus, the cycloalkane yield was enhanced whereas the alkane yield was reduced. Lower aromatics and polynuclear aromatics at a higher temperature gives a very useful insight that rapid coking of catalyst at this temperature should be avoided because the precursors for coke (polynuclear aromatics) are completely suppressed. Earlier work has shown that temperature drastically affected the coke formation during hydrotreatment of BOs.24 Increasing temperature favours the polymerisation for the formation of coke. It was found that at a very high temperature (723 K), the coke formation could be so severe that the reactor was blocked.
 | ||
| Fig. 6 Distribution of types of hydrocarbon (○ alkanes, △ cycloalkanes, ◊ aromatics, and ▽ polynuclear aromatics) in hydroprocessed products for the reaction of 25% BO in GO at various temperatures (H2 pressure of 50 bar, 10 wt% of catalyst, 5 h). | ||
 | ||
| Fig. 7 Distribution of hydrocarbon types (○ alkanes, △ cycloalkanes, ◊ aromatics, and ▽ polynuclear aromatics) in hydroprocessed products for the reaction of 25%, 50% and 100% of BO in GO at 648 K (H2 pressure of 50 bar, 10 wt% of catalyst, 5 h). | ||
Fig. 7 shows that for the hydrocarbon types in hydroprocessed products for 25%, 50% and 100% of BO at 648 K, maximum cycloalkane yield was obtained (19%) for 100% of BO, whereas alkane yield was much less (17%) compared to the other feed percentages of BO at that temperature. In fact, the alkane yield decreased rapidly from feed with 50% BO in GO to feed with 100% BO. Surprisingly, although the aromatics yield was lower (5%) with 100% BO feed, with the polynuclear aromatics yield was rapidly enhanced (by nearly four times) compared to the feeds with 25% and 50% BO. These results indicate that polynuclear aromatics formation by condensation reactions are selectively favored for the 100% BO and this is most likely to be because of its high TAN value obtained via the Diels–Alder reaction and also via secondary reactions of oxygenated compounds such as phenols.25
4. Conclusions
The results indicate that co-hydroprocessing of BO with sulfided CoMoP/Al2O3 catalyst is a promising route for producing transportation fuels. Products obtained from co-processing 25% and 50% of BO with GO contained 2–16% gasoline, 30–35% kerosene, 35–44% diesel, with negligible oxygenates and char. Hydroprocessing of 100% BO at 648 K and a pressure of H2 of 50 bar produced 10% gasoline, 30% kerosene and 30% diesel with a large amount (40%) of undesirable polynuclear aromatics. But at 75 bar (648 K) for 100% BO, polynuclear aromatics formation was suppressed (<2%) and kerosene yield was maximum (41%), with a very small amount of char (1.5%) formation.Acknowledgements
The authors would like to thank the Institute Instrumentation Center (IIC) of IIT, Roorkee, India for the TEM analysis. We would also like to thank the Hydroprocessing Laboratory of CSIR-IIP for help with experimental and analytical work.References
- F. K. Forson, E. K. Oduro and E. Hammond-Donkoh, Performance of Jatropha oil blends in a diesel Engine, Renewable Energy, 2004, 29, 1135–1145 CrossRef CAS.
- A. W. Bhutto, K. Qureshi, R. Abro, K. Harijan, Z. Zhao, A. A. Bazmi, T. Abbas and G. Yu, Progress in the production of biomass-to-liquid biofuels to decarbonize the transport sector–prospects and challenges, RSC Adv., 2016, 6, 32140–32170 RSC.
- M. Asadieraghi, W. M. A. W. Daud and H. F. Abbas, Heterogeneous catalysts for advanced bio-fuel production through catalytic biomass pyrolysis vapor upgrading: a review, RSC Adv., 2015, 5, 22234–32225 RSC.
- J. Wildschut, I. Melian-Cabrera and H. J. Heeres, Catalyst studies on the hydrotreatment of fast pyrolysis oil, Appl. Catal., B, 2010, 99, 298–306 CrossRef CAS.
- J. P. A. Diebold, Chemical and Physical Mechanisms of the Storage Stability of Fast Pyrolysis Bio-Oils: A Review, Colerado, USA, 2000, NREL/SR-570-27613 Search PubMed.
- A. Bridgwater, S. Czernik, J. Diebold, D. Meier, A. Oasmaa and C. Peacocke, Fast Pyrolysis of Biomass: A Handbook, CPL Press, Berkshire, 1999, vol. 1 Search PubMed.
- D. V. Naik, V. Kumar, B. Prasad and M. K. Poddar, Catalytic cracking of Jatropha-derived fast pyrolysis oils with VGO and their NMR characterization, RSC Adv., 2015, 5, 398–409 RSC.
- D. C. Elliott and E. G. Baker, Upgrading biomass liquefaction products through hydrodeoxygenation, Biotechnol. Bioeng. Symp. Suppl., 1984, 14, 159–174 CAS.
- M. F. D. Miguel, M. J. Groeneveld, S. R. A. Kersten, N. W. J. Way, C. J. Schaverien and J. A. Hogendoorn, Production of advanced biofuels: Co-processing of upgraded pyrolysis oil in standard refinery units, Appl. Catal., B, 2010, 26(1–2), 57–66 Search PubMed.
- G. Huber and A. Corma, Synergies between bio- and oil refineries for the production of fuels from biomass, Angew. Chem., Int. Ed. Engl., 2007, 46(38), 7184–7201 CrossRef CAS PubMed.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Liquid-phase catalytic processing of biomass derived oxygenated hydrocarbons to fuels and chemicals, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS PubMed.
- E. Furimsky and F. E. Massoth, deactivation of hydroprocessing catalysts, Catal. Today, 1999, 52, 381–495 CrossRef CAS.
- W. Baldauf, U. Balfanz and M. Rupp, Upgrading of Flash Pyrolysis Oil and Utilization in Refineries, Biomass Bioenergy, 1994, 7, 237–244 CrossRef CAS.
- J. E. Otterstedt, S. B. Gevert, S. G. Jaras and P. G. Menon, Fluid Catalytic Cracking of Heavy (residual) Oil Fractions, Appl. Catal., 1986, 22(2), 159–179 CrossRef CAS.
- M. Anand and A. K. Sinha, Temperature-dependent reaction pathways for the anomalous hydrocracking of triglycerides in the presence of sulfided Co–Mo-catalyst, Bioresour. Technol., 2012, 126, 148–155 CrossRef CAS PubMed.
- P. K. Kanaujia, D. V. Naik, D. Tripathi, R. Singh, M. K. Poddar, L. N. S. K. Konathalaa and Y. K. Sharma, Pyrolysis of Jatropha Curcas seed cake followed by optimization of liquid–liquid extraction procedure for the obtained bio-oil, J. Anal. Appl. Pyrolysis, 2016, 118, 202–224 CrossRef CAS.
- B. S. Rana, R. Kumar, R. Tiwari, R. Kumar, R. K. Joshi, M. O. Garg and A. K. Sinha, Transportation fuels from co-processing of waste vegetable oil and gas oil mixtures, Biomass Bioenergy, 2013, 56, 43–52 CrossRef CAS.
- L. Guoran, L. Wei, Z. Minghui and T. Keyi, Morphology and hydrodesulfurization activity of CoMo sulfide supported on amorphous ZrO2 nanoparticles combined with Al2O3, Appl. Catal., A, 2004, 273, 233–238 CrossRef.
- R. Tiwari, B. S. Rana, R. Kumar, D. Verma, R. Kumar, R. K. Joshi, M. O. Garg and A. K. Sinha, Hydrotreating and hydrocracking catalysts for processing of waste soya-oil and refinery-oil mixtures, Catal. Commun., 2011, 12, 559–562 CrossRef CAS.
- C. Kwak, M. Y. Kim, K. Choi and S. H. Moon, Effect-of-phosphorus-addition-on-the behaviour of CoMoS–Al2O3-catalyst-in-hydrodesulfurization-of-dibenzothiophene-and-4,6 dimethyl dibenzothiophene, Appl. Catal., A, 1999, 185, 19–27 CrossRef CAS.
- J. M. Lewis and R. A. Kydd, The MoO3–Al2O3 interaction: influence of phosphorus on MoO3 impregnation and reactivity in thiophene HDS, J. Catal., 1992, 136(2), 478–486 CrossRef CAS.
- R. D. Back and F. C. P. Grange, Influence of phosphorus on the preparation of CoMo/Al2O3 hydrotreating, Stud. Surf. Sci. Catal., 1998, 118, 517–531 CrossRef.
- M. Gholizadeh, R. Gunawan, X. Hu, F. D. M. Mercader, R. Westerhof, W. Chaitwat, M. M. Hasan, D. Mourant and C. Z. Li, Effects of temperature on the hydrotreatment behaviour of pyrolysis bio-oil and coke formation in a continuous hydrotreatment reactor, Fuel Process. Technol., 2016, 148, 175–183 CrossRef CAS.
- R. Kumar, B. S. Rana, R. Tiwari, D. Verma, R. Kumar, R. K. Joshi, M. O. Garg and A. K. Sinha, Hydroprocessing of Jatropha oil and its mixtures with gas oil, Green Chem., 2010, 12, 2232–2239 RSC.
- M. E. Sanchez, J. A. Menendez, A. Dominguez, J. J. Pis, O. Martinez, L. F. Calvo and P. L. Bernad, Effect of pyrolysis temperature on the composition of the oils obtained from sewage sludge, Biomass Bioenergy, 2009, 33(6–7), 933–940 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra20893b |
| This journal is © The Royal Society of Chemistry 2016 |