
MoO3@Ni nanowire array hierarchical anode for high capacity and superior longevity all-metal-oxide asymmetric supercapacitors†
Abstract
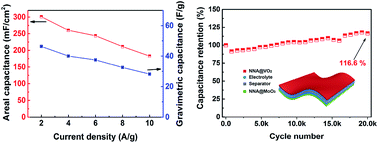
In this communication, a general technology for improving the electrochemical performances of metal oxide based electrode materials has been demonstrated via in situ growth of the electrode materials on Ni nanowire array (NNA) current collector films, which can attain a loading level up to ∼25 mg cm−2. The NNA@MoO3 electrode delivered a high areal capacity ∼477 mF cm−2 and superior longevity (∼5% capacitance loss after 20 000 cycles). Moreover, crystalline VO2 was electro-deposited on NNA as the cathode, which was assembled with the NNA@MoO3 anode into an all-metal oxide-based asymmetric supercapacitor (AAS). The AAS can deliver an open circuit voltage of 1.6 V in an aqueous electrolyte and a high energy density (2.19 mW h cm−3) at the power density of 8.2 mW cm−3. This work provides an example of a flexible AAS device featuring a high areal capacity (307 mF cm−2) and excellent cyclability (116% retention after 20 000 cycles) simultaneously purely involved with metal oxide electrodes.
 Please wait while we load your content...
Please wait while we load your content...

