
Integration of TiO2 photoanode and perovskite solar cell for overall solar-driven water splitting†
 a
a
Abstract
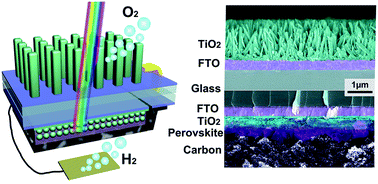
We demonstrate a fully integrated device based on a TiO2 photoanode and perovskite solar cell for overall solar-driven water splitting. The integration substantially broadens the solar absorption spectra and realizes solar water splitting without an external assist. In the integrated configuration, UV light is used by the TiO2 photoanode to generate holes for water oxidation, while the visible light passes through the TiO2 photoanode and irradiates the solar cell. To improve the performance of the integrated device, transmittance of the photoanode is tuned by adjusting the morphology of the TiO2 nanorods, and Sn doping is also introduced to enhance the photoactivity. The solar to hydrogen conversion efficiency is calculated as 1.5%, which is comparable to most of the latest research focused on overall water splitting. Thus, the integrated device provides a promising choice for solar water splitting, considering its high stability under successive illumination.
 Please wait while we load your content...
Please wait while we load your content...

