
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePotential of hybrid iron oxide–gold nanoparticles as thermal triggers for pancreatic cancer therapy
Adeolu Oluwasanmia,
Maryam Malekigorjia,
Stefanie Jonesb,
Anthony Curtisa and
Clare Hoskins*a
aInstitute for Science and Technology in Medicine, School of Pharmacy, Keele University, Keele, ST5 5BG, UK. E-mail: c.hoskins@keele.ac.uk; Tel: +44 (0)1782 734799
bSchool of Life Sciences, Faculty of Natural Sciences, Keele University, Keele, ST5 5BG, UK
First published on 29th September 2016
Abstract
Theranostics are emerging platforms for rapid cancer diagnosis and therapy. Hybrid iron oxide–gold nanoparticles (HNPs) have shown potential as theranostics due to their ability for imaging using MRI, heating using laser irradiation and carrier abilities for drug molecules after surface functionalization. The ability of HNPs to act as localised nano-heaters has been well documented for tumour ablation applications where maximal heating effect is desired. However, the use of HNPs as thermal triggers for drug release requires more control over temperature output and careful consideration of heat dissipation. In this work we report the potential of HNPs to act as localised nano-heaters in vitro and document the cellular effect prior and post laser irradiation in human pancreatic adenocarcinoma (BxPC-3) cell lines. The data showed that after incubation of 50 μg mL−1 HNPs, a thermal increase of up to 9 °C was observed in the cells after laser irradiation with the total area experiencing heat dissipation from the laser beam being 346 mm2. Although the total temperature experienced by cells was below the perceived temperature for irreversible cell damage, after 24 h significant levels of HSP27 and HSP70 were evident with a drop in cell viability to 85%. This indicated that even with rapid irradiation at low temperatures the cells were undergoing stress. Upon I.T. injection in pancreatic xenograft models, a similar heating capacity was observed at identical concentration which also resulted in bulk tumour dissipation. The findings from this work highlight the factors which must be taken into consideration when designing HNPs as theranostics for heat triggered drug delivery.
1. Introduction
Of all cancers, pancreatic cancer is the fourth biggest killer in the western world, with less than 34% of patients surviving 5 years after diagnosis.1 Late diagnosis and treatment reduces the probability of successfully treating the cancer. Often, a delay between diagnosis and treatment exacerbates this. The gold standard treatment for pancreatic cancer is the use of gemcitabine (2,2-difluorodeoxycytidine).2 Gemcitabine is a nucleoside analogue that mimics physiological nucleosides in terms of uptake and metabolism and is incorporated into newly synthesised DNA resulting in synthesis inhibition and chain termination. Unfortunately, this chemotherapeutic agent proves effective in only 23.8% of patients with the only alternative being surgical removal of the localised tumour.3The treatment of pancreatic adenocarcinomas is often hindered by a lack of drug penetration through the dense stroma of the solid tumours formed.4 In order to overcome this problem, higher drug doses are increasingly required in order to attain a therapeutic effect. As a result, patients experience a greater degree and extent of side effects such as increased risk of infection, bruising and bleeding, anaemia, nausea, breathlessness, renal or hepatic impairment, hair loss and tiredness.5 These side effects result from the systemic circulation of the toxic drug agents and their lack of specificity. Nanotechnology is increasingly being exploited for use in cancer therapy with formulations such as Abraxane® and Onivyde® gaining regulatory approval in the last five years.6 The use of nanotechnology is favoured in cancer therapy due to the unique properties of particulates at this size range,7 diversity of architecture and compositions,8 ease of synthesis9 and their ability to passively target cancerous vasculature resulting in deeper tissue permeation via the enhanced permeability and retention effect.10 Advances in nanotechnology for cancer therapy has led to the development of multifunctional entities known as theranostics.
Theranostics are platforms which offer simultaneous diagnosis and therapy, resulting in decreased treatment times.11 These theranostic agents allow for real time imaging after administration, enabling location mapping before initiating therapeutic effect (e.g. ablation/drug release/combination). By coupling treatments to diagnostics and controlling drug release, a rapid and localised clinical effect can be achieved. Early diagnosis in disease states such as pancreatic cancer can mean the difference between life and death. Many types of theranostics have been reported using both organic and inorganic nanoparticulates.12 One class of theranostic agents which has shown potential due to its chemical and physical stability, ease of synthesis and scope for further functionalisation are hybrid nanoparticles.13
Hybrid nanoparticles consisting of an iron oxide core surrounded by a gold shell have shown great promise in cancer therapy.14 Their ability to act as T2 contrast agents for MRI imaging arising from their magnetic iron oxide core and to act as both nano-heaters15 and drug carriers after surface functionalisation of the gold shell has been reported previously.14,16 The use of HNPs as carriers for anticancer agents attached onto the gold surface via linker molecules or with the exploitation of thiol–gold chemistry has been reported.14,16 Wagstaff et al. reported the conjugation of cisplatin onto the surface of HNPs.15 The cisplatin was conjugated through a thiolated polyethylene glycol (PEG) linker. The HNPs produced showed low inherent cytotoxicity on ovarian cancer cell lines (A2780 and A2780/cp70), however the cisplatin conjugated HNPs exhibited up to a 110-fold more cytotoxic effect than the free drug.16 Other studies have been focused on the use of HNPs as drug carriers for pancreatic cancer therapy. Barnett et al. studied the attachment of cytotoxic agent 6-thioguanine (6-TG) onto HNPs.14 In this study, the HNP–6-TG formulation exhibited a 10-fold decrease IC50 in comparison with the free drug in human pancreatic adenocarcinoma (BxPC-3 cells).14
Colloidal gold undergoes surface plasmon resonance (SPR) when irradiated with light of a specific wavelength resulting in the rapid conversion of absorbed light into heat energy.17 The absorbed light energy is rapidly transformed into thermal energy due to the delocalised electrons in the gold shell promoting an increase in temperature; in effect, acting as a localised heating source.18 Hence these HNPs possess the potential to act as nano-heaters for tumour ablation or stimuli responsive drug release.
Studies have highlighted the use of HNPs in thermal ablation of pancreatic cancer. Guo et al. reported cell viability reduction as high as 97.7%, 24 h after irradiation of GoldMag® particles incubated inside human pancreatic (PANC-1) cells.19 Other work has reported the advantages of dual heating HNPs through both laser irradiation and magnetic hyperthermia.20 Espinosa et al. documented the amplification of heating experienced by the HNPs was 15-fold compared with magnetic stimulation only for tumour ablation application. This was tested both in vitro in SKOV3, PC3, and A431 cells lines and in vivo in xenograft skin cancer models.20
The use of HNPs for thermo-responsive drug delivery is now a major focus. This technology benefits from magnetic properties, reduced toxicities and the ability to target the drug release using focused laser irradiation. Previously we reported the controllable heating capacity and resultant dissipation after irradiation of HNPs in agar phantoms at varied concentrations.21 These studies suggested that the HNPs used (50 nm) would be useful for stimuli responsive drug release as their heating could be controlled via alteration of irradiation time and concentration. In order for HNPs to be used to thermally trigger drug release it is important that their temperature rise is not sufficient to cause any thermal degradation of drug or widespread heat dissipation at high temperatures which would affect healthy tissue. As such, for use as a drug carrier and not an ablation agent, it is important that only localised heating is experienced and that any temperatures experienced are not high enough to dissipate away from the tumour site causing unnecessary damage. It is postulated that use of HNPs, which heat up (the bulk) to a maximal temperature below the temperature at which irreversible damage is caused to the cells which is approximately 50 °C, will result in a system that will be most effective.
Studies on the thermal potential and heat dissipation of HNPs for stimuli responsive drug delivery have been reported, however the majority of these have been either been carried out using computer modelling22 or in the use of agar phantoms21,23 and little studies have shown heating abilities after cellular internalization. Whilst gel phantoms are designed to mimic physiological tissue there is an increasing need to fully understand the heating ability and effect on surroundings experienced after irradiation in more realistic conditions either in vitro or in vivo.
In this study the potential of HNPs to act as thermal triggers for drug release is evaluated in vitro using human pancreatic adenocarcinoma cells. The rate of thermal increase and heat dissipation away from the irradiation site will be investigated as well as the effect of HNPs on cells prior to and post irradiation. Additionally the heating effect in solid pancreatic tumours will be mapped post mortem after intratumoural administration in xenograft models.
2. Experimental
2.1 Materials
All chemicals unless otherwise stated were purchased from Sigma Aldrich (UK) and all solvents from Fisher Scientific (UK).2.2 Imaging of HNPs using transmission electron microscopy
HNP suspensions (50 μg mL−1, 2 μL) were dried onto formvar coated copper grids. The grids were imaged using a JEOL JEM-1230 microscope (Jeol, Japan) using anaLYSIS software.2.3 Quantification of cellular uptake of HNPs in BxPC-3 cells
BxPC-3 cells were seeded into 6-well plates (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well) and incubated at 37 °C with 5% CO2. HNPs (5 μg mL−1 & 50 μg mL−1) were incubated with the cells for 24 h. Cells were washed with phosphate buffered saline (PBS) four times and trypsinised. Cells were resuspended (1000000 mL−1) in fresh RPMI media supplemented with 10% foetal bovine serum and 1% penicillin streptomycin (Thermofisher, UK). The cell suspension (1 mL) was placed into an Eppendorf tube and centrifuged at 1000 rpm for 10 min. The supernatant was discarded and the cells were resuspended in 1 mL of concentrated nitric acid. The solutions were incubated at 90 °C for 1 h before dilution in deionised water. The iron (Fe) content was determined using inductively coupled plasma-optical emission spectroscopy (ICP-OES), compared against a calibration of known standards (R2 = 0.999) and the amount of iron internalised was calculated per cell.
000 cells per well) and incubated at 37 °C with 5% CO2. HNPs (5 μg mL−1 & 50 μg mL−1) were incubated with the cells for 24 h. Cells were washed with phosphate buffered saline (PBS) four times and trypsinised. Cells were resuspended (1000000 mL−1) in fresh RPMI media supplemented with 10% foetal bovine serum and 1% penicillin streptomycin (Thermofisher, UK). The cell suspension (1 mL) was placed into an Eppendorf tube and centrifuged at 1000 rpm for 10 min. The supernatant was discarded and the cells were resuspended in 1 mL of concentrated nitric acid. The solutions were incubated at 90 °C for 1 h before dilution in deionised water. The iron (Fe) content was determined using inductively coupled plasma-optical emission spectroscopy (ICP-OES), compared against a calibration of known standards (R2 = 0.999) and the amount of iron internalised was calculated per cell.
2.4 Imaging cellular uptake of HNPs using transmission electron microscopy
Cellular internalisation images were taken after incubation of HNPs with BxPC-3 cells. Cells were grown onto Aclar slides and incubated with HNP (5 μg mL−1 & 50 μg mL−1) for 24 h. Cells were washed with PBS and fixed with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer:2 mM calcium chloride (50:50) for 2 h. The samples were washed for 5 min in 0.1 M sodium cacodylate buffer:2 mM calcium chloride and this step was repeated a three times. The samples were post fixed in 0.1% osmium tetroxide in 0.1 M sodium cacodylate buffer/2 mM calcium chloride for 1 h. A dehydration series was carried out using ethanol before the samples were embedded in spurr resin. The samples were sectioned using a freshly cut diamond knife and placed onto formvar coated copper grids for imaging. The samples were imaged using a JEOL JEM-1230 microscope (Jeol, Japan) using anaLYSIS software.
2.5 Cellular response to HNP exposure using BxPC-3 cells
000 cells per well) and incubated for 24 h. HNPs were incubated with cells (5–500 μg mL−1) for 1, 24, 48 & 72 h at 37 °C and 5% CO2. After incubation, 2 μL of lysis buffer was added to the positive control wells, and the plate was centrifuged at 1500 rpm for 10 min at 37 °C. After centrifugation, 50 μL of the supernatant was removed from each well and placed into a new plate, and 50 μL of a membrane integrity assay reagent was added to the wells. The plates were incubated for 10 min at 37 °C in the dark. Stop reagent (25 μL) was added to the wells, and the LDH levels were recorded at excitation wavelength 560 nm & emission wavelength 590 nm, using a Tecan Pro200 microplate reader (Tecan, UK). The percentage of cytotoxicity was calculated in respect to control wells containing no nanoparticles.000 cells per well) and incubated for 24 h. HNPs were incubated with cells (5–500 μg mL−1) for 1, 24, 48 & 72 h at 37 °C and 5% CO2. Cells were washed with PBS and trypsinised and resuspended in 1 mL fresh media. Cell suspension (50 μL) was mixed with 50 μL trypan blue solution for 1 min. Cells were counted using a Countess® automated cell counter (Thermofisher, UK). Cell viability was determined in respect to control cells with no nanoparticles present.2.6 Investigation into heat generation and dissipation of HNPs in vitro
BxPC-3 cells were seeded into 6-well plates (50000 cells per well) containing quartz coverslips (Alfa Aesar, USA) and incubated at 37 °C with 5% CO2. HNPs (5 μg mL−1 & 50 μg mL−1) were incubated with the cells for 24 h. The cells were washed with phosphate buffered saline (PBS) four times and the coverslips were removed from the plate. The cells were irradiated at 1064 nm using a ML-LASER-YB5 Q-switched Nd:YAG Laser Treatment System (WeiFang MingLiang Electronics Company Ltd., China). Pulse width: 10 ns, pulse repetition frequency: 6 Hz, laser spot diameter: 3 mm, cooling system: water cooled with airflow cooling. The beam was collimated through concave lenses to a 1 mm diameter and passed through the cell sample. The cells were irradiated for 60 seconds and their changing temperature (ΔT) was monitored using Optris PI640 Thermal Imaging Camera (Optris, Germany). The camera was positioned 5 cm away from the cells and focussed on the irradiation site. The data was recorded on Optris PI Connect software (Optris, Germany). Data samples were recorded every 0.1 second and data analysis was carried out offline using direct measurement of the .vga files collected. The ΔT was calculated as per eqn (1):|
ΔT = (Tfinal − Tinitial) − TΔcontrol
| (1) |
Heat dissipation of the samples was measured using pixel analysis offline with optical resolution of 640 × 480 pixel. Effect of multiple irradiation cycles on cellular heating was investigated using 60 seconds irradiation and 60 second cooling cycles repeated over 600 seconds and monitored as above.
2.7 Measurement of heat shock protein production after laser irradiation
BxPC-3 cells were seeded into a quartz 96-well plate (Alfa Aesar, USA) (15000 cells per well) and incubated at 37 °C with 5% CO2. HNPs (5 μg mL−1 & 50 μg mL−1) were incubated with the cells for 24 h. Cells were washed with phosphate buffered saline (PBS) four times and the liquid aspirated from the wells. The cells were irradiated at 1064 nm as previously described for 60 seconds. After irradiation fresh cell culture medium (100 μL) was placed into the wells and the plate was incubated for 24 h.
2.8 Cytotoxicity after laser irradiation
BxPC-3 cells were seeded into 6-well plates (50000 cells per well) containing quartz coverslips (Alfa Aesar, USA) and incubated at 37 °C with 5% CO2. HNPs (5 μg mL−1 & 50 μg mL−1) were incubated with the cells for 24 h. Cells were washed with phosphate buffered saline (PBS) four times. The coverslips were removed from the wells and cells were irradiated at 1064 nm as previously described (60 seconds and multiple irradiation cycles). After irradiation the coverslips were placed back into the wells and fresh media was added and the cells were incubated for 24 h. After this time the cytotoxicity was measured using trypan blue exclusion as previously described. Cytotoxicity was measured in relation to control cells with no nanoparticles.
2.9 Detection of apoptosis via measurement of caspase-3 activity after laser irradiation
BxPC-3 cells were seeded into 6-well plates (50000 cells per well) containing quartz coverslips (Alfa Aesar, USA) and incubated at 37 °C with 5% CO2. HNPs (5 μg mL−1 & 50 μg mL−1) were incubated with the cells for 24 h. Cells were washed with phosphate buffered saline (PBS) four times. The coverslips were removed from the wells and cells were irradiated at 1064 nm as previously described (60 seconds and multiple irradiation cycles). After irradiation the coverslips were placed into the 6-well plate and fresh media was added. The cells were incubated for 24 h. After this time the level of apoptosis was measured using a caspase-3 assay. After 24 h, the supernatant was removed and detached using trypsin. The cell suspensions were centrifuged for 10 minutes at 250 g. The supernatant was removed and 25 μL of cold lysis buffer was added to the cell pellet. Caspase-3 substrate (5 μL, Ac-DEVD-pNA) was added to the samples before transferring to a 96-well plate. The plate was incubated at 37 °C for 1 h before absorbance measurement at 405 nm. Apoptosis levels were measured in relation to control cells with no nanoparticles.
2.10 Imaging of cellular topography after laser irradiation using atomic force microscopy
BxPC-3 cells were seeded into 6-well plates (50000 cells per well) containing quartz coverslips (Alfa Aesar, USA) and incubated at 37 °C with 5% CO2. HNPs (5 μg mL−1 & 50 μg mL−1) were incubated with the cells for 24 h. Cells were washed with phosphate buffered saline (PBS) four times and the coverslips removed from the plate. The cells were irradiated at 1064 nm as previously described. After irradiation the coverslips were returned to the 6-well plates and fresh media was added. The cells were incubated for 24 h. After this time the media was removed and cells washed four times with PBS. The cells were fixed using 2.5% glutaraldehyde in PBS for 10 min. Fixed cells were washed five times with PBS and mounted on glass slides. Cell topography was imaged using a Bruker Catalyst Atomic Force Microscope (Bruker, Germany) using Scan Asyst Adaptive Mode. Cell membrane topography was imaged using a Scan Asyst in Air tip of spring constant 0.4 N m−1, carrying out 896 scans per line at a scan rate of 0.32 Hz and 1.102 V amplitude.
Cell membrane roughness was measured using the topography images with Nanoscope Analysis software (Bruker, Germany). Small areas (1000 × 1000 nm) were chosen at random on ten areas of each cell and their membrane roughness determined. An average was calculated from a total of thirty areas from three cells.
2.11 In situ measurement and imaging of tumour heating in pancreatic xenograft models
Female Nu/Nu mice, five weeks of age (n = 3), (Charles River, UK) were kept in pathogen-free conditions (weight of mice was 20–25 g). All procedures and animal care were carried out according to Project License PPL 70/8806 granted by the UK Home Office. Human pancreatic cancer cell line BxPC-3 was cultured to 90% confluence in RPMI 1640 supplemented with 10% fetal bovine serum and 1% penicillin streptomycin. The cells were washed twice with cold PBS and harvested with trypsin for 10 min at 37 °C. The cells were washed three times with PBS and resuspended in 50:50 media:PBS. The tumour cell suspension (3.0 × 106 cells in 100 μL of 50:50 PBS:media) was injected subcutaneously (s.c.) in the right flank of each mouse. When the tumour became palpable (approximately after one week), measurements in two dimensions with vernier calipers were carried out twice a week and volume tumours calculated according to eqn (2).| V = 4/3π[(D1 + D2)/4]3 | (2) |
Once tumour volume reached a maximum of 0.9 cm3 the mice were sacrificed and used for imaging immediately post mortem. HNP solution (0.2 mg kg−1, 100 μL) was injected intratumorally (I.T.) using a 26 gauge needle (Vet-Tech, UK). The tumour was irradiated at 1064 nm as previously described for 60 seconds and temperature change & heat dissipation measured using an Optris PI640 Thermal Imaging Camera (Optris, Germany). The ΔT data was calculated in relation to a tumour bearing mouse after irradiation with no nanoparticles (only PBS) injected I.T. as per eqn (1).
3. Results and discussion
3.1 Imaging of HNPs using transmission electron microscopy
In this work HNPs composed of an iron oxide core surrounded by a gold shell with attached PEG were used. The HNPs were spherical in shape and 50 nm (±5 nm, n = 25) as observed using TEM (Fig. 1A). The synthetic pathway was monitored using zeta potential measurement, UV-Vis spectrometry and the mass composition ratio of the HNPs was quantified by inductively coupled plasma-optical emission spectroscopy and calculated to be 1:0.3:0.16 (Fe:Au:PEG) as previously described.13 All subsequent concentrations quoted will be based on Fe content of the HNPs.
 | ||
| Fig. 1 Characterisation of HNPs (A) size and morphology carried out using transmission electron microscopy (TEM) & cellular uptake in BxPC-3 cells after 24 h incubation carried out using (B) inductively coupled plasma-optical emission spectroscopy (n = 3 ± SD) and (C) TEM after incubation with 5 μg mL−1 and 50 μg mL−1. | ||
3.2 Cellular uptake of HNPs into BxPC-3 cells
The nanoparticles were incubated with BxPC-3 cells and their cellular uptake measured using ICP-OES. Here the Fe content of the cells was representative of HNP uptake and compared with the physiologically present Fe in control cells. Fig. 1B shows that cellular uptake of the HNPs was concentration dependent with the 50 μg mL−1 sample internalizing 6.9-fold more Fe per cell than the 5 μg mL−1 sample. These findings were in agreement with the TEM images showing HNP uptake (Fig. 1C1 and 2) where more HNPs were observed in the cells incubated with higher concentrations.3.3 Cellular response to HNP exposure
In order for HNPs to be a clinically viable platform for theranostics or drug carriers, they must themselves be relatively non-cytotoxic. In order to investigate the cellular response after exposure and internalization of these particles the cell membrane integrity and cytotoxicity was measured over a 72 h period. Cell membrane integrity was estimated via quantification of LDH release from cells. The data showed that over the 72 h study no significant release of LDH was detected in comparison to the untreated control cells up to 500 μg mL−1 (Fig. 2A). This suggested that the cell membrane had not been compromised upon exposure to the nanoparticles and that nanoparticle uptake was not driven by membrane permeation. It is reported that for the majority of nanoparticles, uptake is driven by endocytosis.24–26 In this work endocytosis is the most likely uptake mechanism as the TEM image (Fig. 1C2) shows nanoparticle accumulation inside what appears to be an endosome under formation. This finding is in agreement with previous more in depth studies regarding cellular uptake of similar HNP structures.13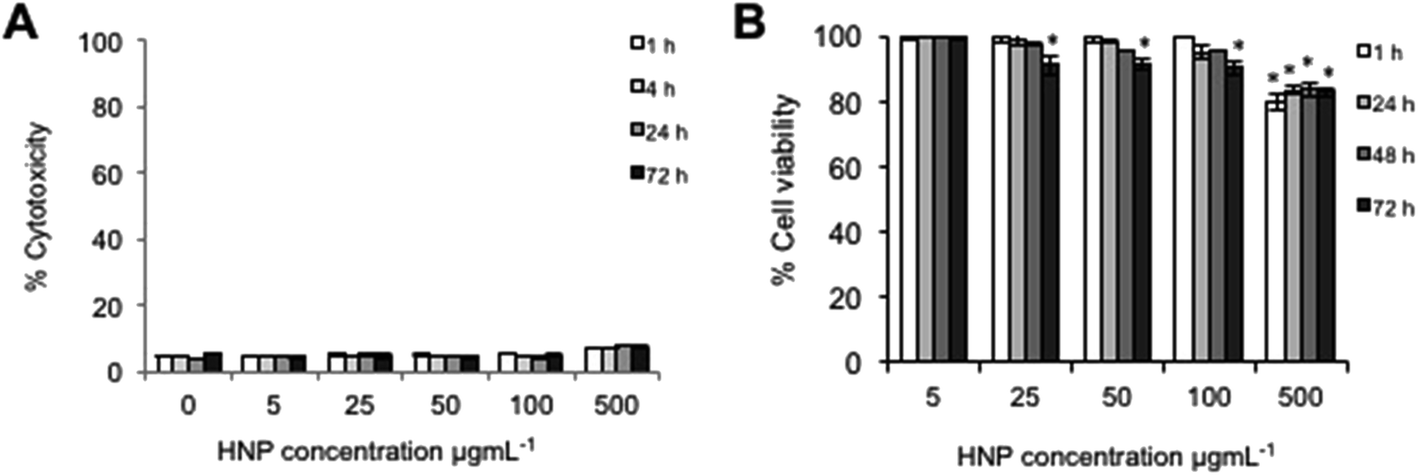 | ||
| Fig. 2 Cellular response of BxPC-3 cells after incubation with HNPs (0–500 μg mL−1) over 72 h. (A) Cell membrane integrity measured via lactate dehydrogenase level & (B) cytotoxicity measured by trypan blue exclusion. All data quoted in relation to control cells (n = 3 ± SD). * denotes significant reduction in viability compared with control sample (p < 0.01). | ||
Cell viability was measured using trypan blue exclusion where a live/dead cell count was measured (Fig. 2B). This study indicated that cytotoxicity was both time and concentration dependent. The data suggested that up to concentrations of 100 μg mL−1 no significant reduction on cell viability was experienced up to 48 h (p > 0.01 although at 72 h a non-significant reduction was evident above 25 μg mL−1). At the highest concentration tested (500 μg mL−1) a significant decrease in cell viability was observed after only 1 h with 80% cell viability (p < 0.01), however, the viability did not further decrease over time. Both the cell membrane integrity and the cytotoxicity studies indicated that at 5 μg mL−1 and 50 μg mL−1, no significant adverse effect to the cells would be experienced due to the presence or internalization of the HNPs.
3.4 Investigation into heat generation and dissipation of HNPs in vitro
Evaluation into the extent of thermal rise experienced in vitro after irradiation of HNPs inside BxPC-3 cells was carried out. The measurements theoretically should give a better indication of potential of the HNPs to act as nano-heaters in biomedicine compared with studies carried out in phantoms. The cells were grown on quartz to ensure no heating effect was experienced due to laser absorption of the coverslips, which would have occurred with glass. The cells were then incubated with HNPs of 5 μg mL−1 & 50 μg mL−1 for 24 h to mimic the cellular uptake quantifications studies. The cells were irradiated using 1064 nm laser light, as previously described13 the UV-Vis absorbance for these particles is very broad with a λmax of 670 nm. However, irradiation is achievable at 1064 nm due to this broad absorbance. Pulsed laser systems operating at 1064 nm are commonly utilized in the cosmetics and ophthalmic industries and hence these systems are reliable and relatively inexpensive. This is an important factor in the translation of such therapies into the clinic. After irradiation, our data showed that at the lower incubation concentration (5 μg mL−1), no major heating effect was experienced with a ΔT of 0.5 °C after 60 seconds (Fig. 3A). However, at the higher concentration of 50 μg mL−1 the HNPs did experience localised heating resulting in a bulk ΔT of approximately 9 °C after the full irradiation time. In contrast to previous work using HNPs suspended in agar phantoms, the thermal rise profile looked remarkably different.21 Previously we found that the HNPs heated up rapidly reaching their maximum temperature (40 °C, 50 μg mL−1) within the first 10 seconds after which a plateau was reached. However, in this study the HNPs appeared to undergo a continual heating profile with their maximum achievable temperature being more than 4-fold less reached after the full 60 seconds of irradiation. This could be due to the fact that the concentrations used in this study are much reduced (not all HNPs incubated become internalised and some will be washed away), by the fact that they are existing in a different, more complex media (presence salts, pH's etc.) or that the heat capacity and ability to dissipate heat inside cells differs from the agar phantoms. In fact, when looking at the cellular uptake study, the concentration of Fe internalised after incubation with 50 μg mL−1 was approximately 26 pg per cell. Looking at the lower concentration incubation (5 μg mL−1), the uptake achieved was approximately 5 pg per cell. This shows that there is a non-linear relationship in the cell internalisation of the particles. After internalisation the cells were estimated to be in the magnitude of 106 in the wells. Hence, it appears that the majority of the HNPs were internalised at the lower concentration incubation. However, at the higher concentration, approximately 50% of total HNPs have been internalised, this is probably the major factor in the difference between phantom and in vitro thermal rise temperatures. | ||
| Fig. 3 Effect of in vitro laser irradiation of HNPs (5 μg mL−1 & 50 μg mL−1) in BxPC-3 cells determined by (A) thermal rise at irradiation point and (B) heat dissipation away from laser focal point measured using a thermal imaging camera (n = 3 ± SD) and (C) temperature of irradiation point over multiple irradiation cycles. | ||
The heat dissipation from the laser focal point was determined after 60 seconds irradiation. Fig. 3B shows the heat dissipation profile for 50 μg mL−1 HNPs incubated in the cells, where a thermal rise was observed after 60 seconds irradiation as far as 10 mm either side of the irradiation point, thus making the total area experiencing a thermal increase 346 mm2. Interestingly, in the phantom studies the heat dissipation at 50 μg mL−1 was detectable using an identical measurement technique at distances only up to 6 mm away from the irradiation point with a total area thermally effected of only 133 mm2. The increase in heat dissipation shown in the in vitro experiments may give rise to the exponential ΔT profile of Fig. 3A. If the irradiation area is dissipating the heat to a greater surrounding area then the HNPs may be losing heat more rapidly and the maximum possible heating temperature for this concentration may not have been reached within the 60 second period and hence no plateau observed.
The effect of multiple irradiation cycles on the heating of HNPs was evaluated using cycles of 60 seconds irradiation followed by 60 seconds rest. This pattern was repeated over 5 cycles to a total of 600 seconds (10 minutes). The heating pattern shown in Fig. 3C shows that the heat generation from laser irradiation using these HNPs was consistent with identical heating and cooling profiles across the timeframe. This indicated that the heat generation did not result in any adverse effects such as damage to the structural integrity of the particles as this undoubtedly would have resulted in a shift in the heating profile.
3.5 Measurement of HSP27 & HSP70 production after laser irradiation
It is important if HNPs are to progress further into clinical applications, that the effect they induce on cells after irradiation is understood. The HNPs themselves have shown no significant toxicity or impairment to the membrane integrity at the two concentrations over the time period being tested (5 μg mL−1 & 50 μg mL−1) as shown in Fig. 2. For application as theranostics or drug carriers in cancer therapy, the heat induced by the HNPs can be harnessed for either tumour ablation, or as a drug release stimuli or indeed a dual platform for both. In this case, it is important to identify how these short sharp irradiations may impact on the cellular state. Hence the production of heat shock proteins (HSPs) was monitored. HSPs are upregulated in response to stress such as temperature changes and exposure to UV radiation. Cells produce HSPs to try and protect themselves from any irreversible damage arising from the stress factor,27 however some studies have suggested that HSPs may aid chemotherapies by acting as chaperones to ensure the drugs reach their desired site in the cell.28For pancreatic adenocarcinoma it is reported that the main HSPs produced after the cells experience thermal increase are HSP27 and HSP70. The measurement of HSP27 with and without irradiation was compared with control cells without irradiation (Fig. 4A). Control cells with no particles were also irradiated with no deviation in temperature or heat shock protein production. The data suggested that the cells incubated with 5 μg mL−1 HNPs did not result in any significant increase in HSP27 production after 24 h (p > 0.01). This was not unexpected as the ΔT profile at this concentration did not show any notable thermal rise after irradiation. However, at 50 μg mL−1 the cells which had been irradiated showed an increase in HSP27 production up to 130% compared with the cells experiencing no irradiation (p < 0.01).
 | ||
| Fig. 4 Cellular response after laser irradiation of HNPs (5 μg mL−1 & 50 μg mL−1) in BxPC-3 cells determined by ELISA measurement of heat shock protein production (A) HSP27 and (B) HSP70 24 h post 60 second irradiation. (C) measurement of cell viability using trypan blue exclusion and (D) apoptosis detection via caspase-3 enzymatic activity measurement 24 h post ■ 60 second and multiple ■ irradiation cycles (n = 3 ± SD) (□ cells incubated with HNPs experiencing no laser irradiation). * denotes significant increase in HSP production compared to control cells, ** denotes significant reduction in viability compared with control sample, *** denotes significant increase in caspase-3 activity compared with control cells (p < 0.01). | ||
Similarly the ELISA for measuring HSP70 (Fig. 4B) showed no significant increase in HSP70 after irradiation with 5 μg mL−1 HNPS but 135% production was observed at 50 μg mL−1 (p < 0.01). These findings show that whilst the total irradiation time of the cells was only 60 seconds and the ΔT was a relatively modest 9 °C compared with previous phantom studies, this was still enough to induce cellular stress.
3.6 Cell viability after laser irradiation
The cell viability was measured 24 h after laser irradiation of cells incubated with 5 μg mL−1 & 50 μg mL−1 HNPS in order to determine if the heat produced resulted in adverse effects resulting in cell death. Fig. 4C shows the % viability of the treated cells with and without laser irradiation compared with untreated control cells. Here the data is in line with the HSP production. Whereby, at 5 μg mL−1 HNP incubation and irradiation, no adverse effect was observed on cell viability. However, a small reduction in viability after 60 seconds laser irradiation was observed at 50 μg mL−1 HNP incubation and irradiation with 85% of cells being viable after the 24 h period (p < 0.01). Interestingly, the cells did not experience any major reduction in cell viability after multiple irradiation cycles compared with the single irradiation samples. This indicates that the cells could be multiply irradiated with little effect to cells and that the temperature itself is the only contributing factor to the cellular fate and not the duration of the cycles. In the case of this study, therefore, the HSPs produced may be adequate enough to allow recovery of cells and hence ablation is not achieved.3.7 Detection of apoptosis via measurement of caspase-3 activity after laser irradiation
Caspase-3 activity was measured as an indication of the apoptotic state of the cells (Fig. 4D). In line with the cell viability, those cells which underwent laser irradiation after incubation with 5 μg mL−1 HNPs did not experience a significant increase in caspase-3 activity. This indicated that the cells were in a healthy state and were not undergoing apoptosis. In contrast, those cells which were incubated with 50 μg mL−1 HNPs did possess significant caspase-3 activity with a 5% increase compared with the controls. These findings were in agreement with the heat shock protein production at higher HNP concentrations after laser ablation. In common with the cytotoxicity, the caspase-3 levels did not increase with increasing irradiation cycles, and hence it can be further confirmed that the temperature alone was sufficient to initiate the changes to cellular state and not necessarily the duration of irradiation.3.8 Imaging of cellular topography after laser irradiation using atomic force microscopy
In order to further investigate the cellular response after laser irradiation of the HNPs the cellular topography was imaged using AFM (Fig. 5). Here the cells were fixed after 24 h post irradiation. The images showed (Fig. 5B2) that cellular morphology was not altered after 60 seconds laser irradiation of HNPs incubated at 5 μg mL−1 compared with the control cells and those with HNPs without irradiation (Fig. 5A1 and B1 respectively). However, in comparison to the control cells (Fig. 5B3) the cells incubated with HNPs at 50 μg mL−1 did have an effect on morphology. Here the cells appeared a little rougher and flatter compared to the control sample, and the cells incubated with HNPs without irradiation and (Fig. 5A1 and B1 respectively). Although the cell viability assays indicated only slight reduction in viability was observed, the AFM images indicate that the cells have undergone some alteration compared with their normal state as a consequence of laser irradiation of the internalised HNPs. However, this did not appear to be exacerbated further upon multiple irradiation cycles, with the cells (Fig. 5C3) looking similar to the single irradiation cells. | ||
| Fig. 5 Effect of HNP irradiation on BxPC-3 cell topography measured using atomic force microscopy. Topography imaged in (1) control cells, (2) cells incubated with 5 μg mL−1 HNPs & (3) cells incubated with 50 μg mL−1 HNPs both (A) untreated and (B) irradiated for 60 seconds and (C) after multiple irradiation cycles. Cells were fixed 24 h post irradiation and imaged using Scan Asyst Adaptive Mode. (D) Roughness analysis carried out from topography images and analysed using NanoScope Analysis software (n = 3 ± SE). □ cells experiencing no irradiation, ■ 60 seconds irradiation, and ■ multiple irradiation cycles. * denotes a significant increase compared to control cells (p < 0.01). | ||
Cell roughness analysis was carried out on the topography images obtained. The data (Fig. 5D) indicated that the cells experiencing the laser irradiation with 50 μg mL−1 HNPs possessed significant increase in membrane roughness compared to the control cells, in agreement with the visual images.
3.9 In situ measurement and imaging of tumour heating in pancreatic xenograft models
The in vitro studies confirmed that once under physiological conditions and at concentrations which may be clinically relevant, the HNPs exhibited different thermal behavior to previous gel phantom studies. To further elucidate the heating potential inside biological systems the HNPs were irradiated after I.T. injection into subcutaneous xenograft pancreatic tumours, volume = 0.9 cm3 (Fig. 6A) immediately postmortem and imaged using the thermal camera. Fig. 6 shows the heating effect on the tumour tissue after 60 seconds irradiation. In the control mouse, no tissue heating effect was observed at the irradiation site (Fig. 6B1–3). In contrast the mouse with HNPs injected I.T. showed localized heated at the laser irradiation point which spread to the entire tumour over the 60 second period. Interestingly, the tumour tissue achieved ΔT of up to 10 °C which is an increase compared to the in vitro studies (although not significant p > 0.01) (Fig. 6D1). However, the heating profile was similar to the agar phantom studies, this perhaps indicated when the HNPs are in more confined areas that their thermal efficiency increases. The fascinating images show that the heat spread or dissipation is confined to the three dimensional tumour structure and does not appear to have dissipated through to the main anatomy of the mouse. In common with the in vitro studies (and contrast to the phantom studies) the HNP irradiation resulted in a thermal increase which could be detected up to 11 mm in each direction from the laser focal point – tumour measurements (11.9 mm × 11 mm). This resulted in an irradiation area of 491 mm2 (Fig. 6D2). After irradiation, the tumours were re-measured to see whether heating of the HNPs had any effect on total tumour volume. This study showed that no difference in tumour volume was experienced after irradiation and that average tumour volume was 0.9 cm3. The relatively increased area experiencing heat dissipation compared to the agar samples indicates that these phantoms may act as indicators of heating potential, but do not indicate in vivo fate. | ||
| Fig. 6 Effect of in situ laser irradiation of HNPs injected I.T. in pancreatic xenograft models. (A) Nu/Nu xenograft (BxPC-3) tumor bearing mouse, (B & C) heat dissipation maps of tumours absent (B) and with 50 μg mL−1 HNPs I.T. (C) at (1) t = 0 s, (2) t = 30 s and (3) t = 60 s. (D) Quantification of (1) thermal rise at irradiation point and (2) heat dissipation away from laser focal point measured using a thermal imaging camera (n = 3 ± SD). | ||
4. Conclusion
With the findings from this work it can be concluded that the HNPs are capable of acting as nano-heaters after laser irradiation at 1064 nm at 50 μg mL−1, which appeared to be an applicable concentration clinically as the particles were well tolerated by the cells. The lower concentration of 5 μg mL−1 did not appear to be effective for nano-heating applications, probably because the particles were too far apart spatially in order for their gold shell heating to adequately cause any major increase in bulk temperature. The production of heat shock proteins in the 50 μg mL−1 HNP irradiation sample indicated that the short duration of irradiation and spike in temperature was adequate to result in cellular stress and this must be taken into account when designing these particles for future application. In this study the cells were incubated at 37 °C and a ΔT of 9 °C resulting in temperatures up to 46 °C. Indeed, there was a small effect on cell viability after irradiation, however, for application in tumour ablation either the irradiation period would need to be extended or the HNP concentration increased to induce a great enough stress on cells resulting in programmed cell death. This correlates with previous studies where they have stated that temperatures as high as 50 °C are required for irreversible cellular damage to occur. However, in vivo it has been reported that cancerous tissue exists at higher temperatures than normal body tissue with temperatures as high as 44 °C, therefore, these HNPs may be sufficient in reaching temperatures of up to 53 °C causing ablation. However, this is yet to be explored. As such, it is suggested that the HNPs in this form (size, morphology, concentration) may be useful in theranostic systems whereby MRI guidance is inferred from the magnetic iron oxide core as well as the heat harnessed from laser irradiation for stimuli responsive drug release. It is postulated that the combined use of drug therapy and in vitro/in vivo heating will result in a synergistic effect resulting in a more efficient rate of kill. This work is currently being investigated.Conflict of interest
The authors have declared that no competing interests exist.Acknowledgements
This work was carried out and funded by the School of Pharmacy and the Institute for Science and Technology in Medicine, Faculty of Medicine and Health Sciences, Keele University. In vivo work was carried out in the School of Life Sciences, Faculty of Natural Sciences, Keele University.References
- A. Jemal, A. Thomas, T. Murray and M. Thun, Ca-Cancer J. Clin., 2002, 52, 23–47 CrossRef PubMed.
- H. Igarashi, T. Ito, T. Hisano, N. Fujimori, Y. Niina, M. Yasuda, T. Kaku, S. Matsuo, T. Oono, M. Yoshinaga, H. Sakai and R. Takayanagi, Case Rep. Oncol., 2011, 4, 534–541 CrossRef PubMed.
- A. Sa Cunha, C. Rault, C. Laurent, X. Adhoute, V. Vendrely, G. Béllannée, R. Brunet, D. Collet and B. Masson, J. Am. Coll. Surg., 2005, 201, 359–365 CrossRef PubMed.
- M. Malekigorji, A. D. M. Curtis and C. Hoskins, J. Nanomed. Res., 2014, 1, 1 Search PubMed.
- J. Wang, X. Zhang, Y. Cen, X. Lin and Q. Wu, Colloids Surf., B, 2016, 146, 707–715 CrossRef CAS PubMed.
- D. Ansari, B. Tingstedt, B. Andersson, F. Holmquist, C. Sturesson, C. Williamsson, A. Sasor, D. Borg, M. Bauden and R. Andersson, Future Oncol., 2016, 12, 1929–1946 CrossRef CAS PubMed.
- E. C. Dreaden, L. A. Austin, M. A. Mackey and M. A. El-Sayed, Ther. Delivery, 2012, 3, 457–478 CrossRef CAS.
- T. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. Yang and Y. Xia, Angew. Chem., 2014, 53, 12321–12364 Search PubMed.
- T. Malarvizhi, M. Remya, M. Ashokumar, K. Muthukumaran, G. Anusha, M. N. Priyadharshni and A. Kiruba, International Journal of Innovative Research and Creative Technology, 2015, 1, 117–123 Search PubMed.
- K. Greish, Methods Mol. Biol., 2010, 624, 25–37 CAS.
- K. Y. Choi, G. Liu, S. Lee and X. Chen, Nanoscale, 2012, 4, 330–342 RSC.
- Z. Fan, P. P. Fu, H. Yu and P. C. Ray, J. Food Drug Anal., 2014, 22, 3–17 CrossRef CAS PubMed.
- C. M. Barnett, M. Gueorgieva, M. R. Lees, D. J. McGarvey and C. Hoskins, J. Nanopart. Res., 2013, 15, 1706 CrossRef.
- C. M. Barnett, M. Gueorguieva, M. R. Lees, D. J. McGarvey, R. J. Darton and C. Hoskins, J. Nanopart. Res., 2012, 14, 1170 CrossRef.
- A. J. Wagstaff, S. D. Brown, M. R. Holden, G. E. Craig, J. A. Plumb and R. E. Brown, Inorg. Chim. Acta, 2012, 393, 328–333 CrossRef CAS.
- X. Huang and M. A. El-Sayed, J. Adv. Res., 2010, 1, 13–28 CrossRef.
- O. A. Yeshchenko, N. V. Kutsevol and A. P. Naumenko, Plasmonics, 2016, 11, 345–350 CrossRef CAS.
- S. Jain, D. G. Hirst and J. M. O'Sullivan, Br. J. Radiol., 2012, 85, 101–113 CrossRef CAS PubMed.
- Y. Guo, Z. Zhang, D. Kim, W. Li, J. Nicolai, D. Procissi, Y. Huan, G. Han, R. R. Omary and A. C. Larson, Int. J. Nanomed., 2013, 8, 3437–3446 CrossRef PubMed.
- A. Espinosa, R. D. Corato, J. Kolosnjaj-Tabi, P. Flaud, T. Pellegrino and C. Wilhelm, ACS Nano, 2016, 10, 2436–2446 CrossRef CAS PubMed.
- A. D. M. Curtis, M. Malekigorji, J. Holman, M. Skidmore and C. Hoskins, J. Nanomed. Nanotechnol., 2015, 6, 6 Search PubMed.
- S. K. Cheong, S. Krishnan and S. H. Cho, Med. Phys., 2012, 36, 4664–4671 CrossRef PubMed.
- C. Hoskins, Y. Min, M. Gueorguieva, C. McDougall, A. Volovick, P. Prentice, Z. Wang, A. Melzer, A. Cuschieri and L. Wang, J. Nanobiotechnol., 2012, 10, 27 CrossRef CAS PubMed.
- A. M. Bannuah, D. Vllasaliu, J. Lord and S. Stolnik, Mol. Pharm., 2014, 11, 4363–4373 CrossRef PubMed.
- N. Oh and J.-H. Park, Int. J. Nanomed., 2014, 9, 51–63 Search PubMed.
- L. Kou, J. Sun, Y. Zhai and Z. He, Asian J. Pharm. Sci., 2013, 8, 1–10 CrossRef CAS.
- S. Fulda, A. M. Gorman, O. Hori and A. Samali, Int. J. Cell Biol., 2010, 214074 Search PubMed.
- G. Jego, A. Hazoumé, R. Seigneuric and C. Garrido, Cancer Lett., 2013, 28, 275–285 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2016 |