
Cadmium(ii) carboxyphosphonates based on mixed ligands: syntheses, crystal structures and recognition properties toward amino acids†
 *a
*a
Abstract
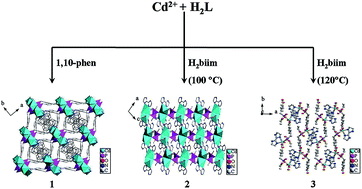
Three novel cadmium(II) carboxyphosphonates with three-dimensional (3D) frameworks and supramolecular structures, namely, Cd3[(L)2(1,10-phen)1.5]·H2O (1), Cd[(L′)(H2biim)] (2), and Cd[(L′)(H2biim)(H2O)]·H2O (3) (L = OOC–C6H4–CH2PO3, L′ = OOC–C6H4–CH2PO2(OC2H5), 1,10-phen = 1,10-phenanthroline, H2biim = 2,2′-biimidazole), were synthesized under hydrothermal conditions and were structurally characterized. For compound 1, the Cd(1)O6, Cd(2)O4N2, Cd(3)O2N2, Cd(4)O4 and CPO3 polyhedra were interconnected in a one-dimensional (1D) chain along the b-axis via corner- and edge-sharing. The adjacent chains were connected with each other by sharing the L3− anions, giving rise to a 3D framework structure. Compound 2 adopted a 3D supramolecular structure. The interconnection of two Cd(1)O4N2 polyhedra and two CPO3 tetrahedra via corner-sharing formed a structure unit, with such units further connected in the bc-plane by sharing the L′2− anions to exhibit a 2D layer, and then the neighboring layers were further assembled into a 3D supramolecular structure by π–π stacking interactions. In compound 3, the Cd(1)O4N2 and CPO3 polyhedra were interconnected to form a unit, and then the interconnection of multi-units were assembled into two kinds of 1D chains, whereby chains and other chains are further connected by π–π stacking interactions to present a 3D supramolecular structure. The luminescence properties of compounds 1–3 were investigated. Meanwhile, the excellent abilities of compounds 2 and 3 for the selective recognition of tryptophan (Trp) were demonstrated.
 Please wait while we load your content...
Please wait while we load your content...

