
A simple and highly diasteroselective approach for the vicinal dichlorination of functional olefins†
Abstract
Organocatalytic stereospecific vicinal dicholorination of a wide variety of functionalized olefins such as ketoesters, esters, ketones, carvone, cholesterol and ethyl sorbate (27 examples) was achieved using inexpensive sulfuryl chloride as well as a simple phosphine catalyst under mild reaction conditions. The products were obtained with good to excellent yields and diastereoselectivities (up to 96% yield and >25 : 1 dr).
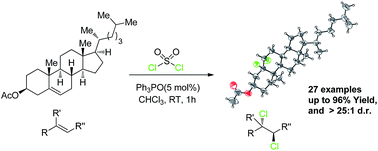
 Please wait while we load your content...
Please wait while we load your content...

