
New insights into catalytic pyrolysis mechanisms and reaction pathways of urea pyrolysis on V–Ti catalyst surfaces†
Yaolin Wang‡
,
Xinbo Zhu‡,
Yu Huang,
Chenghang Zheng and
Xiang Gao*
State Key Laboratory of Clean Energy Utilization, Zhejiang University, Hangzhou 310027, China. E-mail: xgao1@zju.edu.cn
First published on 2nd November 2016
Abstract
A series of V–Ti catalysts with different vanadium loadings for urea pyrolysis were synthesized using the impregnation method and showed desirable catalytic activity. The results showed that the catalysts could accelerate the pyrolysis of urea and inhibit the formation of byproducts. The urea conversion over the catalysts was in the order of 1% V–Ti > 0.5% V–Ti ≈ TiO2 > 5% V–Ti > 10% V–Ti > pure urea, which was associated with the total acidity of the catalysts. The identification and quantification of major byproducts, e.g. biuret, cyanuric acid and melamine, were conducted by HPLC and FT-IR between 100 °C and 450 °C. The reaction pathways of urea pyrolysis in the presence of 1% V–Ti catalysts were proposed based on the byproduct distributions. In the presence of the catalysts, urea pyrolysis was accelerated with a lower initial decomposition temperature. Moreover, the catalysts could promote the further conversion of major byproducts, biuret and cyanuric acid, to the final products NH3 and HNCO at lower temperatures compared to the cases without catalysts.
1. Introduction
Urea is widely used as the reductant in selective catalytic reduction (SCR) of NOx in diesel vehicles due to its unique characteristics including safety, non-toxicity and convenience for transportation.1–4 In commercial urea-SCR systems, urea was initially decomposed into equimolar amounts of ammonia (NH3) and isocyanic acid (HNCO) above the reaction temperature of 134 °C,5 eqn (1). Subsequently, HNCO can be hydrolyzed into NH3 and CO2, eqn (2)6,7Urea pyrolysis:
| CO(NH2)2 → NH3(g) + HNCO(g) | (1) |
HNCO hydrolysis:
| HNCO(g) + H2O → NH3(g) + CO2(g) | (2) |
However, the bottleneck of low ammonia yield and the plugging problem of exhaust pipes and catalyst pores by solid deposits could significantly affect the operation of urea-SCR systems. These phenomena were ascribed to a low urea pyrolysis rate and the formation of various byproducts which can be decomposed only above 400 °C.8–12
Heterogeneous catalysis was proven to be a promising alternative to accelerate urea pyrolysis and improve the NH3 yield of the process. Bernhard studied the promotional effect of various catalyst supports in urea pyrolysis, and the activity followed the order of TiO2 > H-MFI > Al2O3 > ZrO2 > SiO2.13,14 Yang et al. found that one of the zeolites, H–Y, could increase the total yield of NH3 by 28% in the absence of catalysts.15 Eichelbaum illustrated that Fe-beta could accelerated urea pyrolysis, and its rate constants (k) increased from 3.7 × 10−3 (pure urea) to 8.1 × 10−3 (urea/Fe-beta) at 160 °C.16 Yim et al. found that CuZSM5 could enhanced the decomposition of urea solution and increased the NH3 production rate.17 Howard et al. found that commercial SCR catalyst could accelerate the urea decomposition by elimination the second stage of pyrolysis in TGA results, and he roughly pointed out that the urea pyrolysis would undergo a different route due to the catalyst.7
The formation of byproducts was inevitable during urea pyrolysis. Urea pyrolysis and the formation of byproducts have been studied in the absence of catalysts to get a better understanding of the reaction mechanisms of urea pyrolysis. It was reported that larger molecular weight compounds were formed, because HNCO was highly reactive.9,18,19 Schaber and Brack et al. studied the overall reaction of urea pyrolysis and analyzed the side-reactions of urea pyrolysis in the absence of the catalysts.5,20 Very few manuscripts clarified the catalytic effects on the inhibition and decomposition of solid byproducts.21 The variations of the byproduct distribution and the pathways of urea pyrolysis over catalysts were still not clearly understood.
V–Ti, as a commercial de-NOx catalyst, is widely used in urea-SCR systems. It could be promising and meaningful to use V–Ti as the urea decomposition catalyst, and the systems would be highly efficient and compacted. This work aims to study the promotional effect of V–Ti catalysts on catalytic pyrolysis of urea. The pyrolysis process of urea and byproducts over the catalysts was studied by thermogravimetry (TGA). The structure properties of the V–Ti catalysts are characterized by N2 adsorption–desorption, X-ray diffraction (XRD) and ammonia temperature programmed desorption (NH3-TPD). Byproduct distributions of urea pyrolysis over the V–Ti catalysts were analyzed using high performance liquid chromatography (HPLC) and Fourier transform infrared (FT-IR) spectroscopy. Moreover, plausible reaction pathways of catalytic pyrolysis of urea over V–Ti catalysts are proposed based on the identified byproducts.
2. Experimental
2.1 Catalyst preparations
Aqueous urea solution (32.5 wt%) for impregnation was prepared by dissolving appropriate amount of urea (CO(NH)2, 99.0%) in deionized water. Ammonium metavanadate (NH4VO3, 99.0%), oxalic acid (C2H2O4·2H2O, 99.0%) and Degussa P25 support were used in this work. V–Ti catalysts with different vanadium loadings of 0.5, 1, 5 and 10 wt% were prepared by impregnation method. Weighted amount of ammonium metavanadate (NH4VO3, 99.0%) and oxalic acid (C2H2O4·2H2O, 99.0%) solution was added into the TiO2 powder, then stirred and aged under ambient conditions. All the samples were dried at 110 °C overnight and followed by calcination at 500 °C for 4 h. The samples were denoted as x% V–Ti (x represented the weight percentage of V loading, e.g. 1% V–Ti). Typically, 1.0 g of the catalyst powder was wetted with the appropriate amount of 32.5 wt% aqueous urea solution (the mass ratio of catalyst to urea is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), and then the mixture was dehydrated at 40 °C for 72 h in a muffle furnace.15 The samples of major byproducts/catalyst were prepared in a similar way in the follow-up study, where the major byproducts included biuret, cyanuric acid and melamine.
:1), and then the mixture was dehydrated at 40 °C for 72 h in a muffle furnace.15 The samples of major byproducts/catalyst were prepared in a similar way in the follow-up study, where the major byproducts included biuret, cyanuric acid and melamine.
2.2 Characterization of the samples
N2 adsorption–desorption experiments were performed using Quantachrome Autosorb-1C apparatus. The specific surface areas of the samples was evaluated by the Brunauer–Emmett–Teller (BET) equation. Powder X-ray diffraction (XRD) patterns were measured on an X-ray diffractometer (Rigaku, D/MAX 2550, Japan) with a Cu-Kα radiation source.Temperature programmed desorption of ammonia (NH3-TPD) was performed using a chemisorption analyzer (Micromeritics, AutoChem II 2920). The samples were pretreated to remove water vapor and impurities at 100 °C for 2 h, and then treated in N2 streams increased to 650 °C at a ramp of 10 °C min−1.
Thermogravimetric analysis (TGA) experiments were performed using TA-Q500 TGA instrument. A series of experiments were performed using 5 mg of samples, and the heating temperature was maintained at 100 °C for 10 min to dry the samples, then the sample cell was heated to 700 °C at a heating rate was 10 °C min−1.
2.3 Catalytic activity measurements
The pyrolysis experiments were performed using 30 mg of the urea/V–Ti samples (the mass ratio of urea to V–Ti catalysts is 1:1) as the experimental group. Meanwhile, 15 mg of pure urea was used as the comparative group. N2 flow of 350 mL min−1 was used as a sweep gas. The inlet and outlet gases were maintained at 100 °C to prevent urea condensation. In the temperature programmed experiments, the heating rate was set at 10 °C min−1 in a temperature range of 80–700 °C. The outlet concentrations of NH3 and HNCO were continually monitored by an FT-IR spectrometer (Gasmet DX-4000). The yields of the reaction products NH3 and HNCO (eqn (3) and (4)) were calculated as the following equations:
 | (3) |
 | (4) |
Urea conversion (Xurea), which was calculated based on NH3 and HNCO production, was the major evaluation index of catalyst activity, as defined in eqn (5).
 | (5) |
To quantify the residual urea and byproducts, such as biuret (Biu), cyanuric acid (CYA) and melamine (Mel), on the V–Ti samples, high performance liquid chromatography (HPLC) analysis21 was applied using Agilent 1260LC instrument equipped with Zorbax Eclipse XDB-C18 column (150 mm × 4.6 mm i.d.). The photodiode-array detector was set to a measuring wavelength of 200 nm. A 10 mM potassium phosphate buffer solution (volume ratio: KH2PO4:K2HPO4 = 1:3.2) adjusted to pH = 7.5 was used as the eluent. The urea residues were obtained by heating with nitrogen purging at temperatures between 100–450 °C and then quickly cooled to room temperature. The initial mass of the samples were measured, followed by calcination at the setting temperatures. Then the treated samples were washed in the aqueous eluent for chromatographic analyses.
The residual solid intermediates on the catalyst surfaces were further characterized by Fourier transform infrared (FT-IR) spectroscopy with a Nicolet 5700 FT-IR spectrometer. Samples were pretreated by pelletized with KBr. Tested samples were pretreated respectively by calcination in N2 atmosphere at temperatures from room temperature to 450 °C.
3. Results and discussion
3.1 Sample characterizations
Textural properties of the V–Ti catalysts are investigated by BET and XRD. As shown in Table 1, the specific surface areas of the catalysts are influenced by V loadings. The total pore volume decreases from 0.362 cm3 g−1 to 0.315 cm3 g−1 with the increase of vanadium loading, and the change of average pore diameter is hardly observed.| Samples | Surface area (m2 g−1) | Total pore volume (cm3 g−1) | Average pore diameter (nm) |
|---|---|---|---|
| TiO2 | 51 | 0.398 | 27.0 |
| 0.5% V–Ti | 48 | 0.362 | 25.9 |
| 1% V–Ti | 47 | 0.353 | 27.7 |
| 5% V–Ti | 48 | 0.312 | 22.5 |
| 10% V–Ti | 34 | 0.315 | 30.8 |
The XRD patterns of urea/V–Ti mixtures are presented in Fig. 1. All the diffraction peaks match well with anatase TiO2 (ICDD PDF# 21-1272). Under the condition that the vanadium loading is lower than 5 wt%, crystalline V2O5 peaks (ICDD PDF# 54-0513) are not found, which indicates that amorphous V exists on the catalyst surfaces. Meanwhile, the urea/V–Ti samples show strong diffraction patterns for urea (ICDD PDF# 08-0822) and this illustrates that the urea presents in the crystalline forms on the catalyst surface at room temperature.
 | ||
| Fig. 1 XRD patterns of series urea/V–Ti samples; (a) TiO2 (P25); (b) urea/TiO2; (c) urea/0.5% V–Ti; (d) 1% V–Ti; (e) urea/1% V–Ti; (f) urea/5% V–Ti; (g) urea/10% V–Ti. | ||
The surface properties of V–Ti catalysts with different V loadings are investigated by NH3-TPD. As shown in Fig. 2a, there are two NH3 desorption peaks in the NH3-TPD profile of each catalyst sample, one at 240–320 °C, and the other above 360 °C. These two peaks corresponded to the weak and strong acid sites, respectively.22 Furthermore, the relative intensities of the total acidity on the catalyst surface are normalized, as shown in Fig. 2b, and the total acidity reach maximum on the 1% V–Ti surface. Meanwhile, the tendency of total acidity basically corresponds to the yields of NH3 and HNCO, as shown in Fig. 3a.
 | ||
| Fig. 2 (a) NH3-TPD profiles of V–Ti catalysts; (b) surface acidic strength with different V loadings. | ||
 | ||
| Fig. 3 (a) Urea conversion, NH3 and HNCO yields from a series of x% V–Ti samples (under 350 mL min−1 N2 sweep flow with a heating rate of 10 K min−1). NH3 and HNCO yields of (b) pure urea, (c) urea/TiO2 sample and (d) urea/1% V–Ti sample. | ||
3.2 Urea pyrolysis on V–Ti catalysts
NH3 and HNCO yields of urea pyrolysis on different catalysts are shown in Fig. 3. Pure urea pyrolysis shows the lowest NH3 yield of 74% and HNCO yields of 52%, respectively. Catalysts effectively increase the NH3 and HNCO yields of urea pyrolysis and the urea conversion decreases in the order of 1% V–Ti > 0.5% V–Ti ≈ TiO2 > 5% V–Ti > 10% V–Ti > pure urea. The highest yields of NH3 and HNCO are obtained on the 1% V–Ti surface. The results of XRD (Fig. 1) show that V2O5 is well spread and exists in the form of monomeric vanadyl species with V loading less than 1 wt%, while polyvanadate species are formed with high V loading over 5 wt%.23 Thus, monomeric vanadium may be helpful to promote the decomposition of urea and inhibit the formation of byproducts. Meanwhile, the yield of HNCO is obviously lower than NH3, which indicates that HNCO is consumed by side-reactions.The NH3 and HNCO concentrations of pure urea, urea/TiO2 and urea/1% V–Ti pyrolysis process are separately shown in Fig. 3b–d. There is a broad peak between 235–252 °C in NH3 curve of pure urea. It can be that NH3 is generated both from urea pyrolysis (eqn (1)) and the decomposition of byproducts (eqn (7a–d) and (8b)). Conversely, a sharp peak is shown at 200 °C in urea/TiO2, also a much stronger one in urea/1% V–Ti (215 °C). These peaks illustrate that there is less side-reactions between 150 °C and 200 °C over the catalysts. HNCO concentration of pure urea pyrolysis shows a fluctuating increase below 260 °C (Fig. 3b), while HNCO curves in Fig. 3c and d show strong peaks between 160–270 °C and a shoulder peak at 233 °C and 250 °C, respectively. This indicates that catalysts could promote the main reaction (eqn (1)) and inhibit the further reactions of HNCO (eqn (6), (7a and b) and (8a)). The HNCO curve of pure urea also has a weak peak between 350 °C and 450 °C, which is due to the decomposition of byproducts.15 The peaks of HNCO are also observed in Fig. 3c and d respectively, and the intensity of the peaks is much stronger and shifts to lower temperatures between 300–350 °C. Hence it can be deduced that the decomposition of byproducts is accelerated due to the catalytic effect. Generally speaking, HNCO release can be divided into two stages, one comes from the urea pyrolysis reaction (eqn (1)), and the other one (350–450 °C in Fig. 3b; 300–350 °C in Fig. 3c and d) is from the decomposition of byproducts.
NH3 and HNCO curves of urea/TiO2 and urea/1% V–Ti show lower urea pyrolysis temperatures and higher decomposition rates than pure urea pyrolysis, which is a proof of the catalytic effect on urea pyrolysis. Moreover, the urea conversion over the catalysts matches well with the strength of the total acidity, as shown in Fig. 2b. 1% V–Ti exhibits the highest total acidity as well as the best catalytic performance towards urea conversion among V–Ti catalysts. Both Brønsted and Lewis acid sites are attributed to the total acidity of the catalysts. Piazzesi et al. found that Lewis acid sites are crucial for the adsorption and deprotonation of HNCO on the catalyst surface,24 and HNCO hydrolysis can be promoted due to Lewis acid sites.15,25 Meanwhile, Brønsted acid sites are beneficial to the formation of NH4+ ions during the urea pyrolysis.16,26
3.3 TGA of urea catalytic decomposition
The TGA/DTG curves in Fig. 4 depict the decomposition behaviors of pure urea, urea/TiO2 and urea/1% V–Ti. The characteristic temperature and performance of the V–Ti samples are shown in Table 2. The urea decomposition process clearly presents three major decomposition peaks by DTG. The DTG curves of urea pyrolysis shift to lower temperature section due to the catalytic effect, and the weight loss rate of the first stage is increased. | ||
| Fig. 4 (a) TGA graphs of the decomposition of urea (initial weight: 5 mg) and of urea/TiO2 and urea/1% V–Ti 1:1 mixtures (initial weight: 10 mg); (b) DTG curves of pure urea, urea/TiO2 and urea/1% V–Ti. | ||
| Sample | Stage 1st | Stage 2nd | Stage 3rd | Stage 4th | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| WLR | Ti | Tm | WLR | Ti | Tm | WLR | Ti | Tm | WLR | Tm | |
| % | °C | °C | % | °C | °C | % | °C | °C | % | °C | |
| a WLR – weight-loss rate; Ti – the initial decomposition temperature; Tm – the maximum weight loss temperature. | |||||||||||
| Urea | 71.4 | 134 | 193 | 11.5 | 216 | 254 | 15.2 | 263 | 320 | 1.4 | 382 |
| TiO2 | 73.6 | 124 | 178 | 11.0 | 199 | 209 | 14.8 | 238 | 300 | — | — |
| 1% V–Ti | 75.3 | 113 | 176 | 11.3 | 196 | 201 | 13.5 | 235 | 287 | — | — |
In the first stage (130–225 °C), a large mass loss is due to urea pyrolysis producing NH3 and HNCO (eqn (1)). The initial decomposition temperatures (Ti) of urea/TiO2 and urea/1% V–Ti are reduced from 134 °C (pure urea) to 124 °C and 113 °C respectively. The maximum weight loss temperatures (Tm) are decreased from 193 °C (pure urea) to 178 °C (urea/TiO2) and 176 °C (urea/1% V–Ti). In addition, 1% V–Ti shows the highest urea pyrolysis rate and weight-loss rate (WLR), 75.3%, among others. Thus, it is indicated that the catalyst with 1 wt% V loading shows the highest catalytic activity in the first stage.
The second and third stages are at a range of 225–260 °C and 265–325 °C respectively. Both of the stages are mainly due to the decomposition of byproducts, which are formed inevitable with higher decomposition temperatures. In these two stages, the weight-loss rates of urea/TiO2 and urea/1% V–Ti are both lower than pure urea due to the less production of byproducts.
Furthermore, a weak peak is observed between 365–396 °C in the DTG curve of pure urea. The peak proves the existence of polymer which is due to the polymerization of byproducts.5 Yet, there is no other peak in urea/TiO2 and urea/1% V–Ti at higher temperatures, and it is deduced that the polymerization and crystallization of byproducts can be effectively inhibited in the presence of the catalysts.
3.4 Catalyst schematic of urea pyrolysis
The presence of residual urea and byproducts, such as biuret (Biu), cyanuric acid (CYA) and melamine (Mel) in samples is analyzed by HPLC and the mass of byproduct distribution is presented in Table S1.† The retention times of the standard substances are 3.182 min (urea), 3.625 min (CYA), 4.320 min (Biu) and 5.879 min (Mel), and each of the standard substances matches well with the HPLC results of the samples respectively. Besides, the characteristic groups on tested samples are detected by FT-IR (Fig. 7). The identification of the weak peak with the residence time of 3.477 min between 200–350 °C was further characterized by FT-IR due to the limitation of HPLC. The peak represents the existence of another byproducts, ammelide (Amme), which leads to low recoveries at the specific temperatures.The results of quantitative analysis of the main components of the tested samples is normalized and shown in Fig. 5. According to the distribution of residual components, urea decomposition process could be divided into three steps as follows.
 | ||
| Fig. 5 The byproduct distribution of the urea pyrolysis (by normalization method); (a) pure urea; urea pyrolysis on (b) TiO2 and (c) 1% V–Ti; (d) the selectivities of HNCO and NH3. | ||
In the first step, urea pyrolysis (eqn (1)) is the main reaction of the step, and large amounts of NH3 and HNCO are formed between 100–200 °C. In the presence of the catalysts, residual urea decreases from 0.54 mol per mol-urea (pure urea) to 0.20 mol per mol-urea (urea/TiO2) and 0.09 mol per mol-urea (urea/1% V–Ti), which indicates that 1% V–Ti can significantly decrease the unreacted urea and increase the urea pyrolysis rate. The FT-IR spectra of urea (Fig. 7a) at room temperature presents two strong peaks (3446 cm−1 and 3345 cm−1), due to the asymmetric stretching vibrations of –NH2. Some shoulders between 3300–3250 cm−1 correspond to the symmetric stretching vibrations of –NH2 and the effect of hydrogen bond. Then they become stronger at 150 and 200 °C due to the polymerization of urea, which has been studied that the polymerization would decrease the yields of NH3 and HNCO.5 However, these features are diminished in the spectra of urea/TiO2 and urea/1% V–Ti (Fig. 7b and c) between 150–200 °C. So it can be deduced that the catalysts could inhibit the polymerization of urea and promote its depolymerization.
In the beginning of the second step (200–350 °C), Biu is the superior byproduct by the further reaction between HNCO and residual urea (eqn (6)) along with the urea pyrolysis. And there is no Biu detected until the temperature above 150 °C and the mass of Biu reaches the maximum at 200 °C without catalysts, while the generation temperatures of Biu decreases below 150 °C in the presence of catalysts. Meanwhile, the decomposition of Biu has not led to a reproduction of CYA between 300–350 °C, which indicates that Biu could directly decomposes into NH3 rather than participating in further reaction above 300 °C.
Biu production:
| CO(NH2)2 + HNCO(g) → NH2–CO–NH–CO–NH2 | (6) |
In the absence of the catalysts, CYA at about 200 °C. And the mass of CYA increases from 0.01 mol per mol-urea to 0.10 mol per mol-urea, while Biu decreases by 0.13 mol per mol-urea in Fig. 6a (pure urea). This indicates that most biuret is converted into CYA by eqn (7d), and partial HNCO may begin to react with biuret to produce CYA (eqn (7a and b)), or produce CYA itself (eqn (7e)). However, on the TiO2 and 1% V–Ti surfaces, the formation of CYA takes place below 200 °C, and the mass of CYA reaches the maximum at 200 °C. It could be deduced that CYA is mainly from the polymerization of HNCO (eqn (7e)), which means the main forming path of CYA has been changed. This has further led to a decline in temperature of the CYA production by 50 °C due to the catalytic effect. The mass of CYA rapidly decreases between 250–300 °C, while it still remains at 0.10 mol per mol-urea without the catalysts at 300 °C. Besides, TiO2 has a little stronger promoting effect than 1% V–Ti according to the residual mass of Biu and CYA, which are also confirmed by TG in Fig. 6a and b respectively.
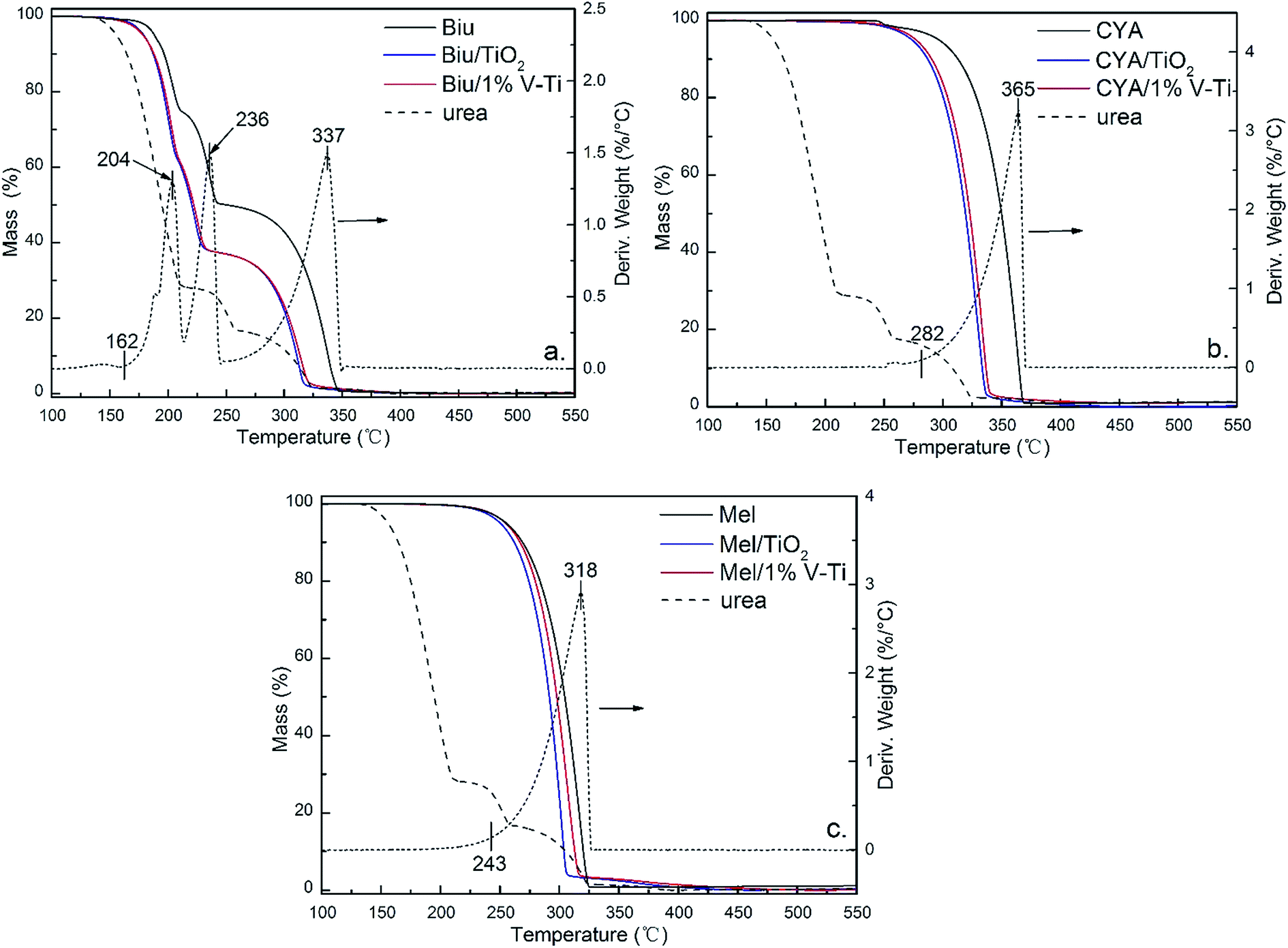 | ||
| Fig. 6 TGA graphs of the decomposition of (a) biuret, (b) cyanuric acid and (c) melamine (initial weight: 5 mg) and of byproducts/TiO2 and 1% V–Ti 1:1 mixtures (initial weight: 10 mg). | ||
CYA production:
| Biuret + HNCO(g) → C3N3(OH)3(CYA) + NH3(g) | (7a) |
| Biuret + HNCO(g) → (triuret) → CYA + NH3(g) | (7b) |
| Biuret + urea → CYA + 2NH3(g) | (7c) |
| 2Biuret → CYA + HNCO(g) + 2NH3(g) | (7d) |
| 3HNCO(g) → CYA(s) | (7e) |
Meanwhile, a small amount of Mel is detected in all samples. Catalysts have little influence on Mel pyrolysis (Fig. 6c) but there is still less Mel on TiO2 and 1% V–Ti because of the catalytic decomposition of Biu and CYA.
Mel production:
| Biuret + HNCO(g) → (ammelide) → (ammeline) → melamine + H2O(g) | (8a) |
| CYA + NH3(g) → (ammelide) → (ammeline) → melamine + H2O(g) | (8b) |
In the second step, the formation of byproducts is obviously reduced due to the catalytic effect so that there are higher yields of NH3 and HNCO over the catalysts. The conclusion is consistent with the results in Fig. 3. The selectivity of NH3 and HNCO are calculated by the residual mass of byproducts, as shown in Fig. 5d. It is found that the selectivity of NH3 and HNCO over TiO2 and 1% V–Ti are higher than the pure urea between 200–350 °C.
Fig. 7 shows FT-IR spectra of solids in urea decomposition. Characteristic signals for urea and its solid decomposition byproducts are summarized in Table S2.† It can be observed that Biu (1330 cm−1 and 1083 cm−1) and CYA (3200 cm−1) are produced at 150 °C over the catalysts, while the characteristic vibrations of Biu and CYA are not observed until 200 °C in pure urea. The characteristic peak of CYA (3197 cm−1) at 350 °C can be detected without catalysts, while it is found below 300 °C on the catalysts. The conclusion of FT-IR spectra can be well in accordance with the results of HPLC.
 | ||
| Fig. 7 FT-IR spectra measured during the process of urea pyrolysis; (a) pure urea; urea pyrolysis on (b) TiO2 surface and (c) 1% V–Ti surface; RT – room temperature; (details shown in Table S2†). | ||
Additional, the weak peaks between 2050–2300 cm−1 on urea curves show the existence of volatile salts of HNCO, ammonium cyanate, [NH4+NCO−], or hydronium cyanate, [H3O+NCO−], for example.5 These peaks vanish above 300 °C. However, there are only several peaks on TiO2 and 1% V–Ti, which indicates that the catalysts could promote the decomposition of volatile salts of HNCO formed during the urea decomposition. The band (1722 cm−1) from the C–O stretching vibration of Amme is started to be observed at 250 °C,27 while this peak is only seen between 200 °C and 300 °C on the catalysts. It is indicated that the formation of other byproducts, such as Amme, can be also reduced due to the catalysts.
Furthermore, the anionic form of adsorbed urea and the (–NCO) group were proven by the peaks at 1562–1552 cm−1 and 2202 cm−1 respectively in the previous studies.28–31 And these characteristic bands are also observed on TiO2 and 1% V–Ti at 200 °C and 250 °C respectively in Fig. 7b and c. It is proposed that Lewis acid sites plays a major role in the formation of the (–NCO) group on the catalyst surface.24 And it is deduced that Lewis acid sites may be helpful to inhibit the formation of cyanate salts, since these sites can bind strongly the anionic form of adsorbed urea and the (–NCO) group.
The main reaction of the third stage (above 300 °C) is the decomposition of the residue. A large proportion of residual urea unreacted would be deposited, and the residue would lead to an ease of redeposition in a vicious circle. The catalysts could effectively reduce the residual urea from 0.06 mol per mol-urea (pure urea) to 0.02 mol per mol-urea (urea/TiO2) and 0.01 mol per mol-urea (urea/1% V–Ti) at 450 °C. In the FT-IR spectra, residual Amme in pure urea is undecomposed at a high temperature, but it is completely decomposed over the catalysts.
In general, a plausible reaction scheme for urea pyrolysis under catalytic condition is shown in Fig. 8. In the absence of the catalyst (1% V–Ti), urea pyrolysis (1) initially takes place at about 134 °C in Table 2, followed by the formation of Biu (2) between 150–200 °C, as shown in Fig. 5a. Then a certain amount of CYA is formed by the transformation of Biu (3) above 200 °C, and its decomposition (4) begins above 300 °C (Fig. 5a). Meanwhile, a little amounts of Mel above 200 °C and Amme above 250 °C are observed in the pure urea by FT-IR spectra, Fig. 7a. However, the overall reaction of urea pyrolysis is accelerated due to the catalytic effect. In the presence of 1% V–Ti, the initial decomposition temperature of urea pyrolysis (1′) decreases by 20 °C (Table 2). Then, the formation temperature of Biu (2′) is declined below 150 °C (Fig. 5c), and Biu can be easily decomposed less than 162 °C (3′) as shown in Fig. 6a. The most prominent change of the catalytic effect is that most of CYA is formed directly from the polymerization of HNCO (2′′) rather than from Biu (3), and this change of the reaction path leads to a lower generation temperature below 200 °C. And the decomposition temperature of CYA also shifts to a lower temperature below 300 °C (4′) over the catalysts in Fig. 5c. Moreover, other byproducts, such as Mel and Amme, are hardly observed in Fig. 7c, especially above 300 °C, and it is deduced that there must be trace residue rather than polymer and crystals on the 1% V–Ti surface.
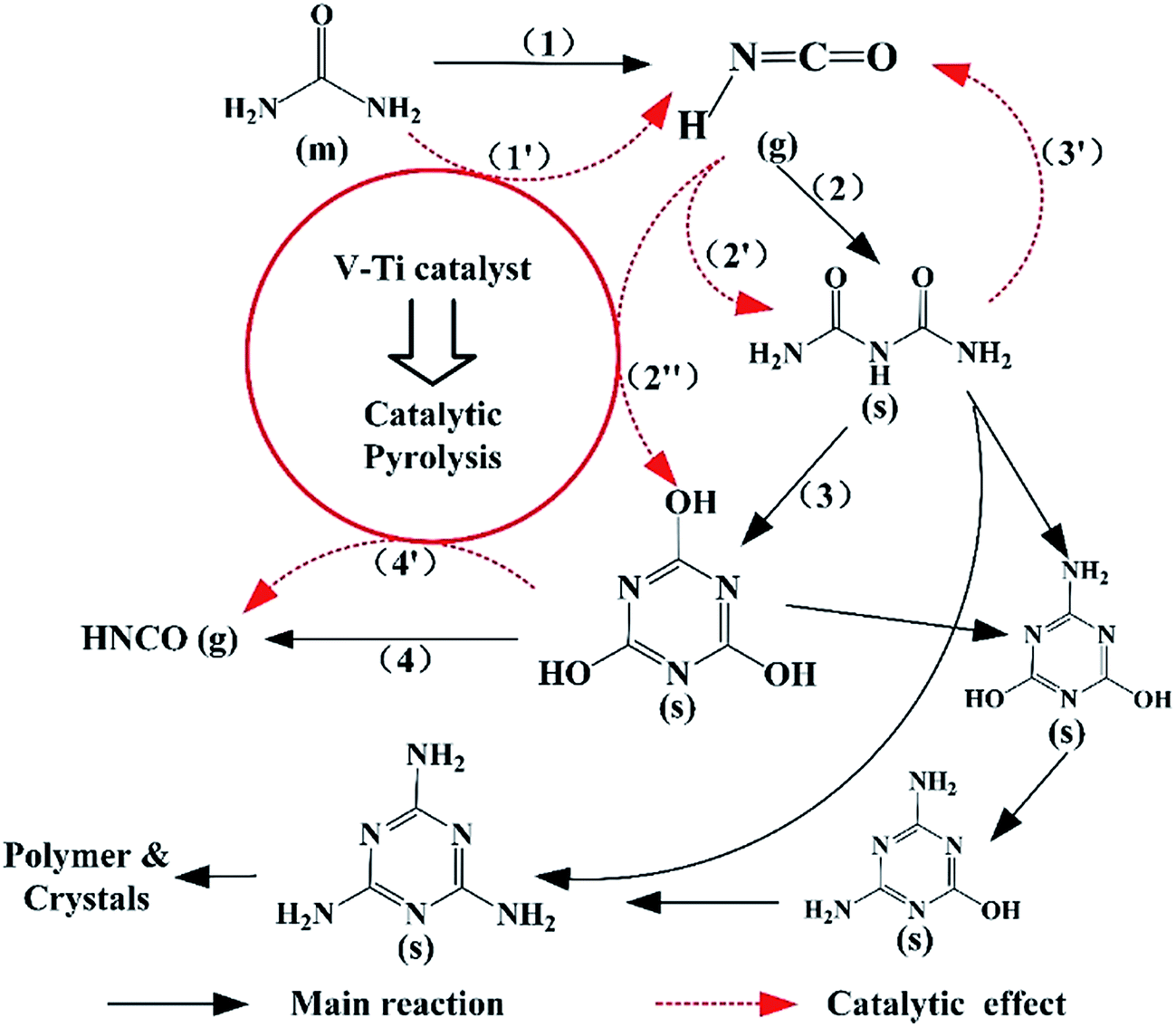 | ||
| Fig. 8 Proposed schematic of the major reactions and byproducts for the catalytic pyrolysis of urea (m – melt; g – gas; s – solid). | ||
4. Conclusion
The urea pyrolysis on a series of V–Ti catalysts (0.5 wt%, 1 wt%, 5 wt%, 10 wt%) at 80–700 °C were experimentally investigated. 1% V–Ti showed the highest urea conversation and reaction selectivity than others, especially the pure urea. The catalytic effect on urea pyrolysis decreased in the order 1% V–Ti > 0.5% V–Ti ≈ TiO2 > 5% V–Ti > 10% V–Ti > pure urea. And the yields of the gaseous product were basically consistent with the total acidity of catalysts influenced by V loading. More acid sites would enhance the adsorption of urea and the (–NCO) group on the catalyst surface. TGA/DTG showed the catalytic effect could accelerate the rate of urea pyrolysis.Moreover, HPLC and FT-IR were applied to measure the byproducts distribution of urea pyrolysis. The amount of the residual urea was declined remarkably due to the catalytic effect. Meanwhile, the main reaction path was changed based on comparing the mass variation of the byproducts. The changed formation path of CYA, eqn (7e), led to a lower reaction temperature, followed by less amount of CYA at 300 °C. It was also found that the pyrolysis of Biu and CYA would be influenced by catalysts (Fig. 5a and b). Further work is under progress to study the adsorption kinetic and mass-transfer of the catalytic pyrolysis of urea and byproducts on V–Ti surface.
References
- J. Li, H. Chang, L. Ma, J. Hao and R. T. Yang, Catal. Today, 2011, 175, 147–156 CrossRef CAS.
- J. C. Wang, Z. L. Peng, Y. Chen, W. R. Bao, L. P. Chang and G. Feng, Chem. Eng. J., 2015, 263, 9–19 CrossRef CAS.
- P. L. T. Gabrielsson, Top. Catal., 2004, 28, 177–184 CrossRef CAS.
- S. Brandenberger, O. Kröcher, A. Tissler and R. Althoff, Catal. Rev., 2008, 50, 492–531 CAS.
- P. A. Schaber, J. Colson, S. Higgins, D. Thielen, B. Anspach and J. Brauer, Thermochim. Acta, 2004, 424, 131–142 CrossRef CAS.
- M. Kleemann, M. Elsener, M. Koebel and A. Wokaun, Ind. Eng. Chem. Res., 2000, 39, 4120–4126 CrossRef CAS.
- H. L. Fang and H. F. M. DaCosta, Appl. Catal., B, 2003, 46, 17–34 CrossRef CAS.
- M. Koebel, M. Elsener and M. Kleemann, Catal. Today, 2000, 59, 335–345 CrossRef CAS.
- M. Koebel and E. A. Strutz, Ind. Eng. Chem. Res., 2003, 42, 2093–2100 CrossRef.
- V. O. Strots, S. Santhanam, B. J. Adelman, G. A. Griffin and E. M. Derybowski, Emissions, 2009, 2, 283–289 Search PubMed.
- L. Xu, W. Watkins, R. Snow, G. Graham, R. McCabe, C. Lambert and R. O. Carter, Sae Technical Papers, 2007 Search PubMed.
- P. Hauck, A. Jentys and J. A. Lercher, Catal. Today, 2007, 127, 165–175 CrossRef CAS.
- A. M. Bernhard, D. Peitz, M. Elsener and O. Krocher, Top. Catal., 2013, 56, 130–133 CrossRef CAS.
- A. M. Bernhard, D. Peitz, M. Elsener, T. Schildhauer and O. Krocher, Catal. Sci. Technol., 2013, 3, 942–951 CAS.
- W. Yang, Z. Chen, J. Zhou, Z. Huang and K. Cen, Ind. Eng. Chem. Res., 2011, 50, 7990–7997 CrossRef CAS.
- M. Eichelbaum, R. J. Farrauto and M. J. Castaldi, Appl. Catal., B, 2010, 97, 90–97 CrossRef CAS.
- S. D. Yim, S. J. Kim, J. H. Baik, I. S. Nam, Y. S. Mok, J. H. Lee, B. K. Cho and S. H. Oh, Ind. Eng. Chem. Res., 2004, 43, 4856–4863 CrossRef CAS.
- L. Stradella and M. Argentero, Thermochim. Acta, 1993, 219, 315–323 CrossRef CAS.
- M. Koebel and M. Elsener, J. Chromatogr. A, 1995, 689, 164–169 CrossRef CAS.
- W. Brack, B. Heine, F. Birkhold, M. Kruse, G. Schoch, S. Tischer and O. Deutschmann, Chem. Eng. Sci., 2014, 106, 1–8 CrossRef CAS.
- A. M. Bernhard, D. Peitz, M. Elsener, A. Wokaun and O. Krocher, Appl. Catal., B, 2012, 115, 129–137 CrossRef.
- D. Nicosia, I. Czekaj and O. Kroecher, Appl. Catal., B, 2008, 77, 228–236 CrossRef CAS.
- G. Busca, M. A. Larrubia, L. Arrighi and G. Ramis, Catal. Today, 2005, 107–08, 139–148 CrossRef.
- G. Piazzesi, O. Kröcher, M. Elsener and A. Wokaun, Appl. Catal., B, 2006, 65, 55–61 CrossRef CAS.
- G. Piazzesi, M. Devadas, O. Kröcher, M. Elsener and A. Wokaun, Catal. Commun., 2006, 7, 600–603 CrossRef CAS.
- G. Piazzesi, M. Elsener, O. Kröcher and A. Wokaun, Appl. Catal., B, 2006, 65, 169–174 CrossRef CAS.
- L. S. Rangel, J. R. de la Rosa, C. J. L. Ortiz and M. J. Castaldi, J. Anal. Appl. Pyrolysis, 2015, 113, 564–574 CrossRef.
- M. A. Larrubia, G. Ramis and G. Busca, Appl. Catal., B, 2000, 27, L145–L151 CrossRef CAS.
- P. Hauck, A. Jentys and J. A. Lercher, Appl. Catal., B, 2007, 70, 91–99 CrossRef CAS.
- A. M. Bernhard, I. Czekaj, M. Elsener and O. Kröcher, Appl. Catal., B, 2013, 134–135, 316–323 CrossRef CAS.
- M. A. Larrubia, C. Ramis and G. Busca, Appl. Catal., B, 2001, 30, 101–110 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra20031a |
| ‡ These authors contributed equally to this work |
| This journal is © The Royal Society of Chemistry 2016 |