
Electrospun hematite nanofiber/mesoporous silica core/shell nanomaterials as an efficient adsorbent for heavy metals†

Abstract
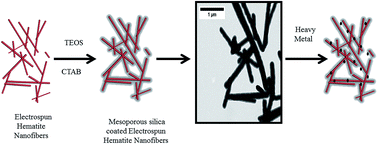
Functionalized nanomaterials hold tremendous promise for water treatment because their high surface area makes them ideal sorbents for pollutants like heavy metal ions that are pervasive in global water supplies. Here, a novel core/shell nanomaterial consisting of an electrospun hematite nanofiber core and a mesoporous silica shell of tunable thickness (from 20–60 nm) was prepared for the first time. The synthesis involved careful control of pH and sequential addition of the silica source to control the growth and ultimately, thickness of the mesoporous silica shell on the electrospun hematite nanofiber. The core/shell structure was subsequently tailored for heavy metal adsorption by grafting an aminopropyl functional group on the mesoporous silica surface. The resulting electrospun hematite/mesoporous silica core/shell nanomaterials were extensively characterized by energy dispersive spectroscopy (EDS) with high resolution transmission electron microscopy (HRTEM), and ζ potential measurements both before and after adsorption of the Cr(III), from aqueous solution. Notably, sorption capacities for Cr(III) exceeded those previously reported for other nanostructured sorbents for this metal. The advantages of these core/shell materials include controllable surface area through introduction of porosity and the option for facile surface modification to optimize physicochemical interactions for pollutant uptake. These nanocomposites also exhibit improved chemical resistance in harsh environments. At acidic pH values, for example, the core/shell nanomaterials were more chemically resistant to iron dissolution than the parent electrospun hematite nanofibers, which broadens the range of waste streams to which these sorbents can be applied.
 Please wait while we load your content...
Please wait while we load your content...

