
Design, synthesis and evaluation of diphenyl ether analogues as antitubercular agents†
Abstract
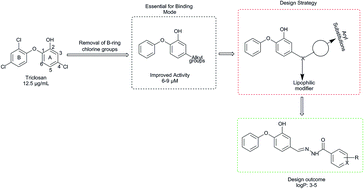
We herein report the investigation of new diphenyl ethers as Mycobacterium tuberculosis enoyl-acyl carrier protein reductase (InhA) inhibitors by structure-based drug design approach. The virtual library of diphenyl ethers was designed and molecules with appreciable physicochemical and ADMET properties were docked. The best ranked molecules based on docking studies were synthesized and characterized by spectral studies. Synthesized compounds were evaluated for in vitro antitubercular activity against Mycobacterium tuberculosis H37Rv strain by Microplate Alamar Blue Assay. Among the tested compounds, DE3 and DE2 exhibited substantial antitubercular potential at 3.125 and 6.25 μg mL−1 concentrations, respectively. The most active compounds were further evaluated for cytotoxicity studies against Vero and HepG2 normal cell lines by microculture tetrazolium assay and ascertained to be safe against normal cell. The molecular dynamic study reveals that the best active compounds show better binding free energy than the reference compounds TCl and JPL at Mtb InhA binding site.
 Please wait while we load your content...
Please wait while we load your content...

