
pH-tuned diverse structures and properties: two Anderson-type polyoxometalate-based metal–organic complexes for selective photocatalysis and adsorption of organic dyes†
Abstract
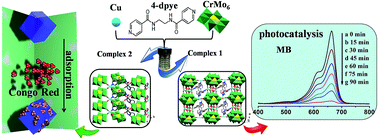
Two novel Anderson-type polyoxometalates (POMs)-based metal–organic complexes, namely, Cu5(μ2-OH)2(4-dpye)2[CrMo6(OH)5O19]2(H2O)10 (1), {Cu(4-Hdpye)[CrMo6(OH)6O18](H2O)2}·2H2O (2) (4-dpye = N,N′-bis(4-pyridinecarboxamide)-1,2-ethane), were hydrothermally synthesized in different pH ranges and structurally characterized by single-crystal X-ray diffraction, IR spectra, powder X-ray diffraction (PXRD) and thermogravimetric analyses (TGA). Complex 1 shows a 3D (3,4)-connected framework constructed by the 2D {Cu5(μ2-OH)2[CrMo6(OH)5O19]2} inorganic layer and bidentate 4-dpye bridging ligands. Complex 2 exhibits a 2D polymeric structure based on the 2D {Cu[CrMo6(OH)6O18](H2O)2} layer and monodentate 4-Hdpye ligands. The complexes 1 and 2 prove that the pH value plays an important role not only in the synthesis and structures but also for the properties of the title complexes. Their electrochemical behaviour and electrocatalytic activities towards the reduction of bromate and hydrogen peroxide have been reported. In addition, the selective photocatalytic properties and adsorption of organic dyes for 1 and 2 have been investigated. Complex 1 possesses good photocatalytic activity towards the degradation of organic dyes Congo Red (CR) and methylene blue (MB), while complex 2 has high adsorption capacity of MB/CR at room temperature. All MB/CR molecules adsorbed on 2 can be completely released in NaCl-containing DMF solution.
 Please wait while we load your content...
Please wait while we load your content...

