
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and NMR analysis of 13C and 15N-labeled long-chain polyamines (LCPAs)†
Maryna
Abacilar
a,
Fabian
Daus
a,
Christian
Haas
a,
Stephan Ingmar
Brückner
b,
Eike
Brunner
*b and
Armin
Geyer
*a
aFachbereich Chemie, Philipps-University Marburg, Hans-Meerwein-Straße 4, 35032, Marburg, Germany. E-mail: geyer@staff.uni-marburg.de
bFachrichtung Chemie und Lebensmittelchemie, TU Dresden, 01062 Dresden, Germany. E-mail: eike.brunner@tu-dresden.de
First published on 26th September 2016
Abstract
15N, 13C, or both isotope labels were introduced in selected positions of several LCPAs containing 5, 11, or 13 nitrogens. The number of transformations during chemical synthesis was minimized by coupling Fmoc-glycine-15N, Fmoc-glycine-1-13C, or Fmoc-β-alanine simultaneously at both ends of the growing oligoamide chain on resin-linked norspermidine. LCPAs containing 10–20 nitrogens are the main organic constituent of diatom biosilica. Preliminary NMR studies of bioinspired silica nanocomposites obtained from double-labeled LCPA 14 and 29Si-enriched orthosilicic acid identified the close spatial arrangement of 29Si and 13C nuclei.
Introduction
Long-chain polyamines (LCPAs) have found considerable research interest in the context of silica biomineralisation. These molecules were found to be tightly attached to the biosilica from diatoms1,2 and sponges.3 LCPAs could even be detected in sedimented diatom silica from the bottom of the oceans.4In vitro experiments revealed that LCPAs are capable of accelerating the precipitation of silica from silicic-acid containing solutions.5 These discoveries have initiated numerous in vitro studies as well as biomimetic silica synthesis experiments.6–11 It is therefore commonly assumed that LCPAs are key molecules for the formation of diatom biosilica. However, the molecular interactions between silicic acid/silica species and LCPAs are not yet fully understood. Solid-state NMR investigations including DNP-enhanced experiments on fully 13C, 15N, and 29Si-labeled diatom biosilica have shown that LCPAs are present at the organic/inorganic interface, i.e., in close contact with the silica.12,13 Solid-state NMR spectroscopy is in principle able to reveal the interactions at the LCPA–silica interface by using distance-sensitive experiments such as rotational echo double resonance (REDOR13–16) and others. This is, however, complicated in fully isotope labeled samples due to signal overlap. Specifically isotope labeled samples are thus highly desirable. The present manuscript describes the synthesis of specifically 13C,15N-labeled LCPAs of similar structure as found in diatoms. It is demonstrated that nanocomposites prepared in vitro from these LCPAs and 29Si-labeled silica exhibit remarkable dephasing when rotor-synchronous π pulses are applied at the Larmor frequency of 29Si. That means, the present synthesis and labeling strategies open new perspectives for the application of advanced solid-state NMR techniques to study the interactions between LCPAs and silicic acid/silica species in detail.Natural LCPAs are microheterogenic mixtures with average chain lengths and variable degrees of methylation. Chemical synthesis allows the incremental variation of LCPA structure and alkylation degree to quantify the markedly different biomineralisation properties of individual LCPA chain lengths.17 Here, we present recent progress on the synthesis of LCPAs and applications for the site-specific labeling of individual nitrogen and carbon atoms.
Results and discussion
The experimental methodology applied for the synthesis of LCPAs has to take into account the increasing basicity of a growing oligoamine chain. Repeated reductive aminations or N-alkylation methods for the assembly of oligoamines containing more than 10 amino groups are hampered by severe analytical problems of the increasingly polar target compounds. Moreover, the analytical purity of synthetic LCPAs is difficult to prove because of non-stoichiometric amounts of TFA and the small 1H NMR chemical shift dispersion of the alkyl groups. In order to avoid polar oligoamine intermediates, we employ a synthetic strategy which evades them before the very last reaction step. For this purpose, we adapted the borane reduction of oligoamides for the synthesis of LCPAs with up to 18 nitrogens.18 The oligoamide precursors are obtained by standard amide coupling methods of solid-phase peptide synthesis and finally reduced on the resin. A test cleavage of the oligoamides from the resin and its analytical proof before the final reduction step is an efficient method to obtain pure LCPAs. The number of chemical transformations is minimized by coupling β-alanine simultaneously on both ends of the growing oligoamide as shown in Fig. 1.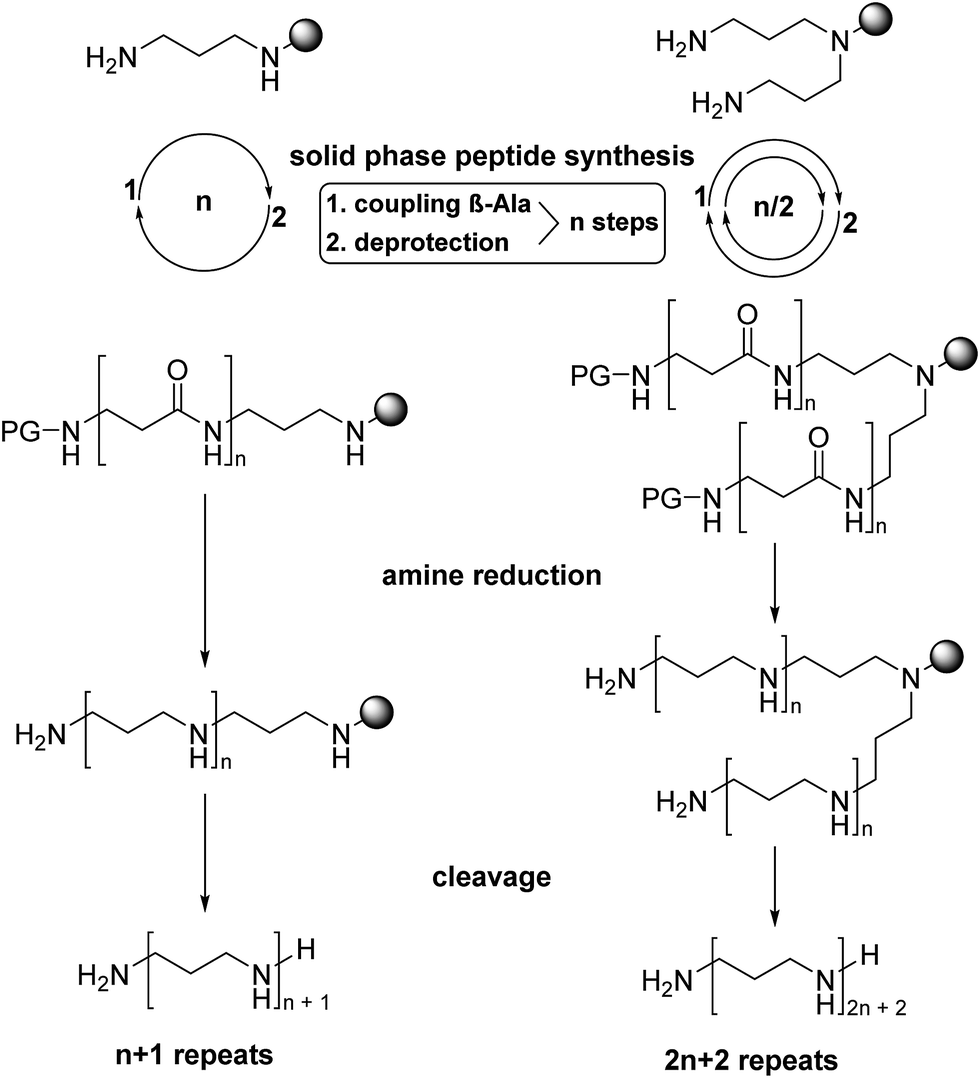 | ||
| Fig. 1 General synthetic strategy. Propylenediamine or norspermidine were linked to 2-chlorotritylchloride resin (grey shaded circle) and elongated by repeated acylation with Fmoc-β-alanine to the oligoamide precursors of the desired length. Borane reduction followed by TFA cleavage yields the polyamines as TFA salts. | ||
The secondary amino group of norspermidine reacts regioselectively with 2-chlorotrityl chloride resin when its two primary amines are protected with the DDE protecting group [DDE = N-(1-(4,4-dimethyl-2,6-dioxocyclohexylidene)ethyl)]. Hydrazinolysis of DDE yields the norspermidine-loaded resin 1a as shown in Scheme 1 (intermediate oligoamides which are linked to the resin are characterized as “a”). The 15N isotope labels are introduced by coupling of resin 1a with Fmoc-Gly-15N to obtain 3a. The test cleavage of a small amount of resin sets free the oligoamide precursor 3b (test cleavages “b” are characterized in the ESI†). Complete borane reduction of 3a finally yields the pentaamine LCPA 4 as a TFA salt after acidic cleavage from resin. The isolated LCPA TFA salts are characterized in the Experimental section.
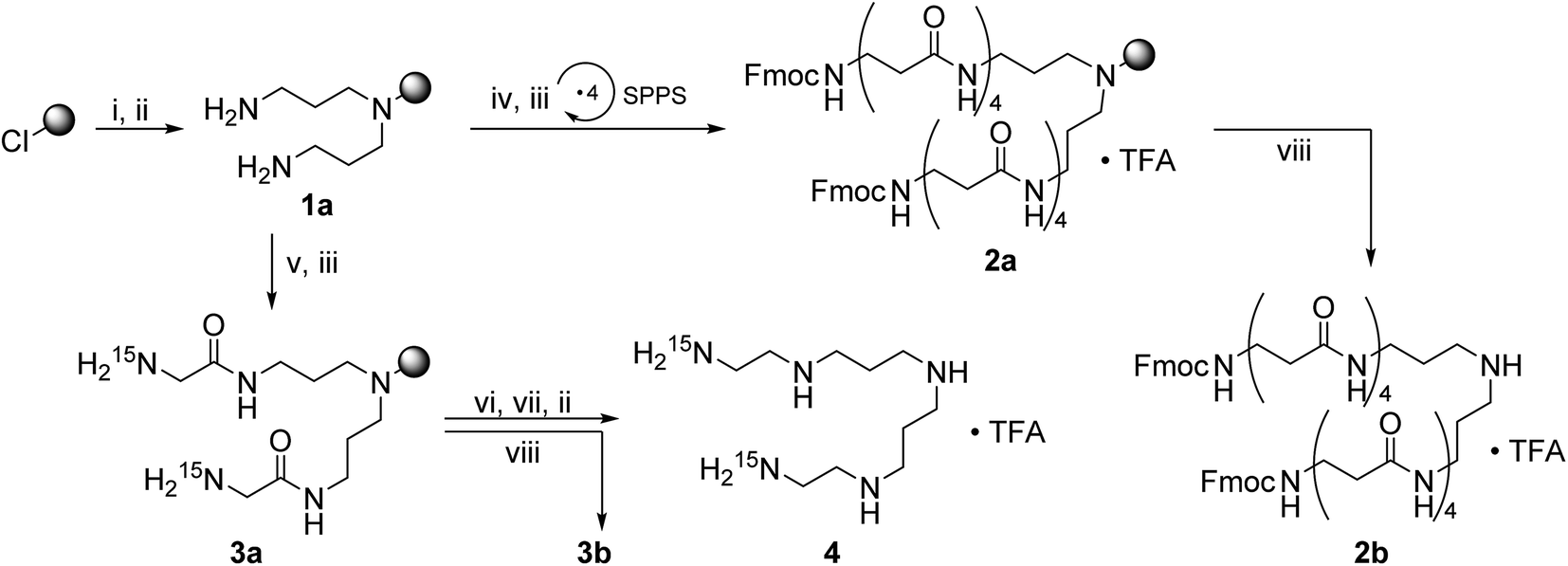 | ||
| Scheme 1 Synthesis of the 15N-labeld LCPA 4 and resin-linked oligoamide 2a. A grey shaded circle represents the 2-chlorotritylchloride resin and a downward arrow with the reaction conditions (viii) the NMR test cleavage. (i) 1,9-Bis-DDE-norspermidine, DIPEA, DCM, rt, 16 h; (ii) 2% hydrazine in DMF, rt, 1 h; (iii) 25% piperidine in DMF, rt, 30 min; (iv) Fmoc-β-Ala, HOBt, HBTU, DIPEA, DMF, rt, 1 h; (v) Fmoc-Gly-15N, HOBt, HBTU, DIPEA, DMF, rt, 1 h; (vi) 1.0 M BH3·THF, 60 °C, 5–7 d; (vii) 25% piperidine in DMF, 60 °C, 2–4 d; (viii) 95% TFA in H2O. | ||
Scheme 2 shows the synthesis of the LCPAs 6, 8, and 10 starting either from loaded resin 1a by automated peptide synthesis on a peptide synthesizer or by manual coupling from 2a. These three LCPAs have the same number of 13 nitrogens. The isotope label on 10 were introduced in form of 13C-enriched glycine. Scheme 3 shows the synthesis of double-labeled LCPAs 12 and 14 which contain two 13C and two 15N labels each but differ in the position of the label within the LCPA chain.
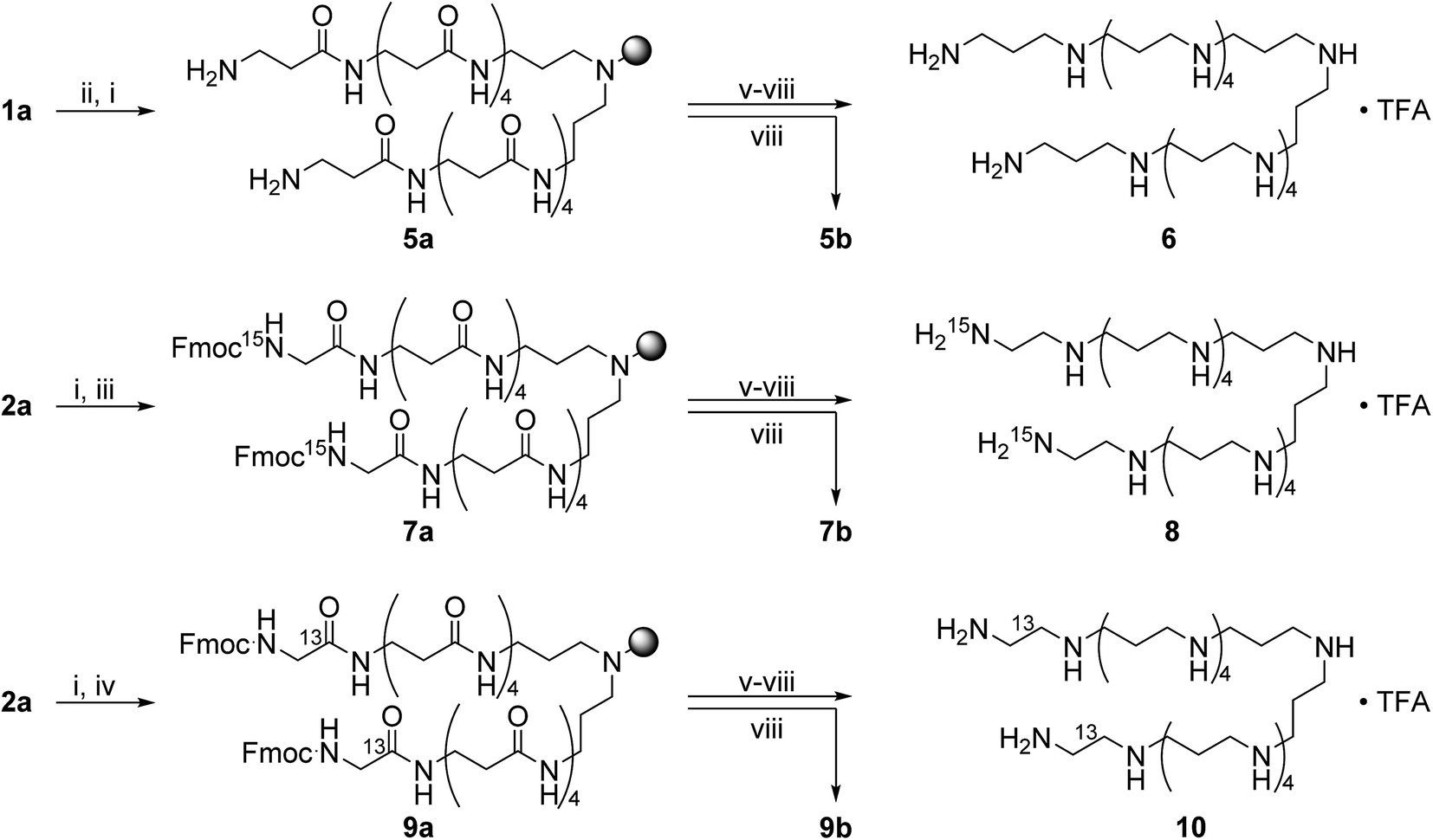 | ||
| Scheme 2 Synthesis of the LCPAs 6, 8, and 10 which are unlabeled or bear a single 13C or 15N label. (i) 25% piperidine in DMF, rt, 30 min; (ii) Fmoc-β-Ala, HOBt, HBTU, DIPEA, DMF, rt, 1 h; (iii) Fmoc-Gly-15N, HOBt, HBTU, DIPEA, DMF, rt, 1 h; (iv) Fmoc-Gly-1-13C, HOBt, HBTU, DIPEA, DMF, rt, 1 h; (v) 1.0 M BH3·THF, 60 °C, 5–7 d; (vi) 25% piperidine in DMF, 60 °C, 2–4 d; (vii) 2% hydrazine in DMF, rt, 1 h; (viii) 95% TFA in H2O. | ||
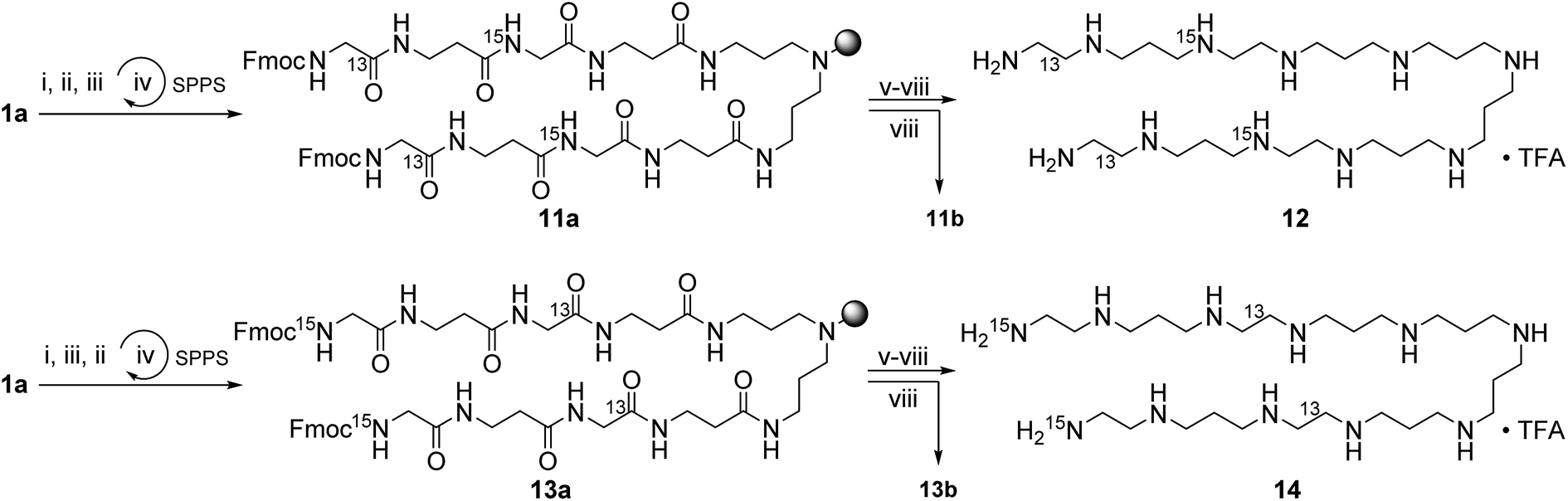 | ||
| Scheme 3 Synthesis of the double-labeled LCPAs 12, and 14. (i) Fmoc-β-Ala, HOBt, HBTU, DIPEA, DMF, rt, 1 h; (ii) Fmoc-Gly-15N, HOBt, HBTU, DIPEA, DMF, rt, 1 h; (iii) Fmoc-Gly-1-13C, HOBt, HBTU, DIPEA, DMF, rt, 1 h; (iv) 25% piperidine in DMF, rt, 30 min; (v) 1.0 M BH3·THF, 60 °C, 5–7 d; (vi) 25% piperidine in DMF, 60 °C, 2–4 d; (vii) 2% hydrazine in DMF, rt, 1 h; (viii) 95% TFA in H2O. | ||
Fig. 2 shows the 13C and 29Si CP MAS NMR spectra of a dried silica gel prepared from fully 29Si-labeled silicic acid in the presence of compound 14. The main 13C signal can be assigned to the isotope label as shown in Scheme 3. The assignment of the signals in the 29Si CP MAS NMR spectrum is given in the figure. The result of a 1H–13C–{29Si}–REDOR experiment is shown in Fig. 3 (left).
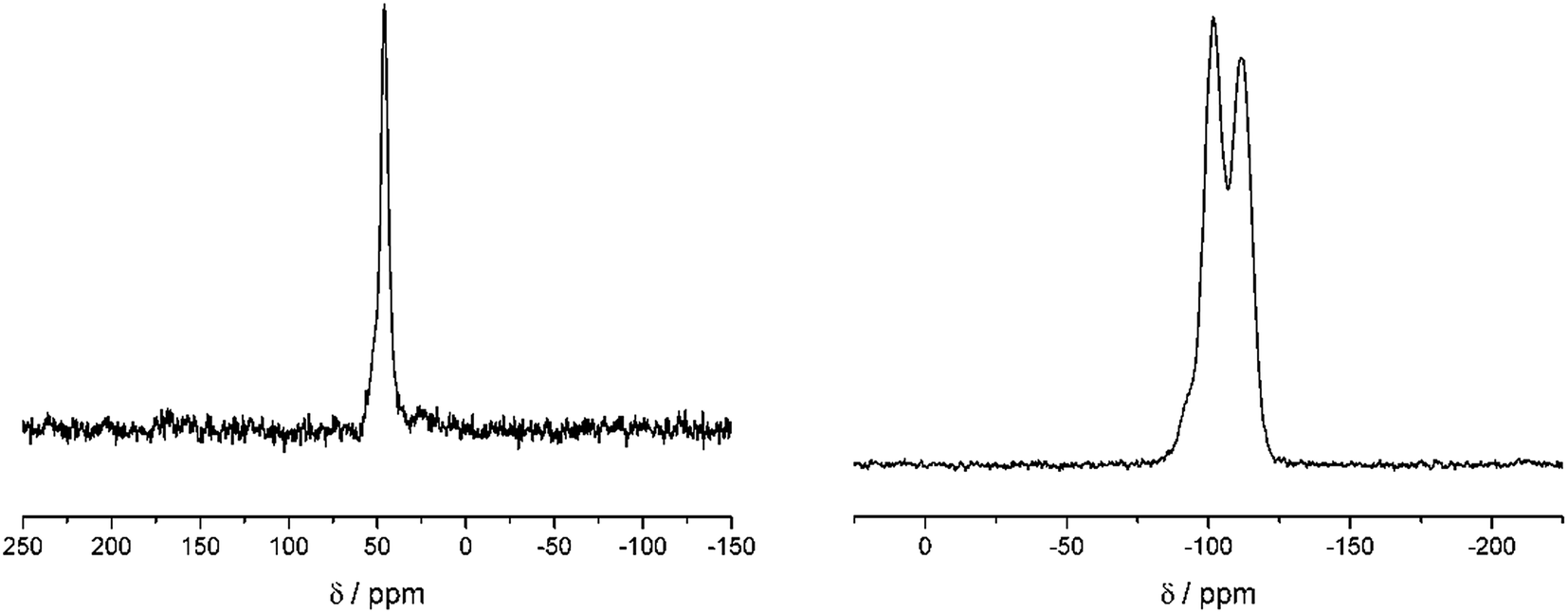 | ||
| Fig. 2 Left: 1H–13C CP Hahn echo spectra of the dried silica gel containing 14 after 2 ms echo time. 2048 scans were acquired. Exponential line broadening set to 50 Hz. Right: 1H–29Si CP Hahn echo of the dried silica gel containing 14 after 10 ms echo time indicating the condensation of Si(OH)4 by the appearance of Q3 and Q4 groups. 6144 scans were acquired. Exponential line broadening was set to 50 Hz. | ||
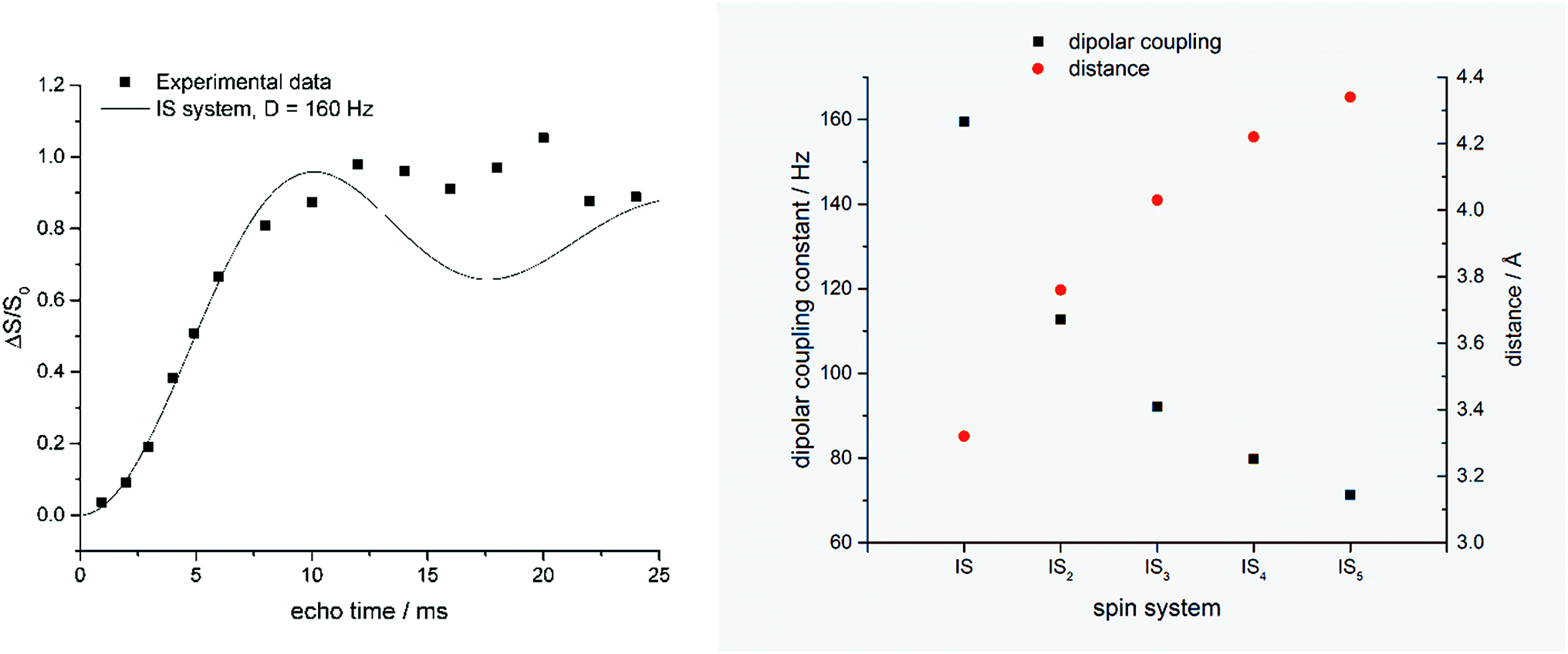 | ||
| Fig. 3 Left: REDOR fraction (ΔS/S0) of the dried silica gel containing 14 measured by 1H–13C–{29Si} CP REDOR. Black line shows simulated curve for a two-spin (IS) system with a dipolar coupling constant of 160 Hz (simulated with Simpson version 4.0.0.20). Right: Relation of internuclear distances and individual IS dipolar coupling constants calculated for equidistant ISn spin systems. | ||
The REDOR fraction Rf is plotted as a function of echo time N × tr, i.e., the product of the rotor period tr and the number of rotor cycles N. The REDOR fraction is defined as ΔS/S0 with ΔS = S − S0. S and S0 are the 13C spin echo amplitudes with and without dephasing pulses for 29Si, respectively. The REDOR fraction increases up to 1 which means that the 13C signal is fully dephased. This remarkably strong REDOR effect indicates a strong 13C–29Si interaction. All 13C labels are obviously in close contact with 29Si in this composite. Analysis of the initial REDOR curve, i.e., for N × tr using the approach described by Bertmer and Eckert19 yields a second moment of 2.0 × 105 Hz2 for the 13C–29Si dipolar interaction. Using the simple two-spin approximation, this corresponds to a 13C–29Si internuclear distance of 3.35 Å. This is unexpectedly low. Furthermore, the two-spin approximation does not describe the measured REDOR curve for long dephasing times. It is, therefore, likely that each carbon label is in contact with more than one 29Si nuclei.
Fig. 3 (right) shows the result of a calculation using the assumption that each 13C nucleus couples with n = 1, 2, 3, or 4 equidistant 29Si nuclei. Further experiments and calculations are necessary in order to obtain a more detailed insight into this spin system. In any case, selectively labeled LCPAs form nanocomposites with silicic acid in which they are in very intimate contact with the condensed silica. It is likely that each 13C label is in close proximity of more than one 29Si.
Conclusion
The assembly of LCPAs with 5, 9, to 13 nitrogens bearing site-specific isotope labels (13C, 15N or double-labeled) was achieved by borane reduction of an oligoamide precursor on chlorotrityl resin. The number of chemical transformations was minimized by coupling β-alanine simultaneously at both ends of the growing oligoamide chain on resin-linked norspermidine. Solid state NMR spectroscopy identified the close spatial arrangement of 13C and 29Si nuclei in biomineralized silica obtained from LCPA 14 and orthosilicic acid. The quantitative NMR analysis of LCPAs containing site-specific labels in different positions elucidates molecular details about the relative orientation of LCPAs and the SiO4 tetrahedron in amorphous silica. Further systematic variations of LCPAs length and labeling position will yield further atomistic details about the borderline between organic and inorganic matter in diatom biosilica and bioinspired silica nanocomposites.Experimental section
General
All chemicals and solvents used for the synthesis of peptide building blocks or peptide synthesis were purchased from commercial sources and were not purified further. The purification of the peptides was achieved by semipreparative reversed-phase HPLC with a Thermo Scientific Dionex UltiMate 3000 with a MWD-3000 detector and a Macherey-Nagel VP Nucleodur C18 gravity column (5 μm, 125 × 21 mm), using the eluents A: H2O + 0.1% TFA and B: MeCN + 0.085% TFA. Afterwards, the peptides were lyophilized with a Christ Alpha 2–4 LDplus. The NMR spectroscopy (1H, TOCSY, HSQC, ROESY, 13C) was performed with a Bruker AV-300 or AV-500/HD-500 spectrometer. High-resolution ESI(+) mass spectra were recorded in the positive mode with a Finnigan LTQ-FT from Fisher Thermo Scientific.Resin loading (1a)
N 1,N9-Bis-1-(4,4-dimethyl-2,6-dioxo-cyclohexylidene) ethyl-norspermidine was prepared from norspermidine (1.0 equiv.) in a reaction with 2-acetyl dimedone (2.0 equiv.) by refluxing in ethanol for 4 h.21 2-Chlorotritylchloride resin (1.6 mmol g−1) was loaded with the double-protected norspermidine (6.0–19.0 equiv.) and DIPEA (6.0 equiv.) in DCM (3.0 mL mmol−1) at rt for 18 h or 22 h. The resin was filtered, washed with DCM and MeOH (3.0 mL mmol−1) two times for 30 min. Finally, the resin was treated several times with a mixture of NEt3/DMF 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, DMF, MeOH, DCM before it was dried under vacuum. Then DDE-groups were removed with 2% hydrazine in DMF (vol%) three times for 30 min. The resin was washed several times with DMF, MeOH, DCM before it was dried under vacuum. The loading was determined by NMR test cleavage.
:4, DMF, MeOH, DCM before it was dried under vacuum. Then DDE-groups were removed with 2% hydrazine in DMF (vol%) three times for 30 min. The resin was washed several times with DMF, MeOH, DCM before it was dried under vacuum. The loading was determined by NMR test cleavage.
NMR resin test cleavage
A small amount of dry resin (1–2 mg) was treated with trifluoroacetic acid (0.1 mL) at ambient temperature. After 2–5 min DMSO-d6 (0.5 mL) were added and the 1H NMR was measured of the solution.Automated solid phase peptide synthesis
Peptide 5a was synthesized on a microwave equipped CEM Liberty Blue automated peptide synthesizer. The resin 1a was swollen in DMF for 30 min at ambient temperature. Then resin 1a was treated with Fmoc-β-Ala (5.0 equiv.), DIC, Oxyma in DMF at 50 °C for 10 min for each active site. Fmoc deprotection was performed with 20% piperidine in DMF for 2.5 min at 50 °C.Manual solid phase peptide synthesis
Peptide 2a was synthesized manually. Firstly, the precursor resin 1a was swollen in DMF for 30 min at ambient temperature under nitrogen atmosphere. Then, the resin was treated with Fmoc-β-Ala (4.0 equiv.), HBTU (4.0 equiv.), HOBt (4.0 equiv.) and DIPEA (4.0 equiv.) in DMF two times for 60 min for each reactive groups. Fmoc deprotection was achieved with 25% piperidine in DMF for 10 min and 20 min followed by the same washing steps.Coupling of isotope-labeled Fmoc-Gly
The precursor resin 2a was swollen in DMF for 30 min at ambient temperature under nitrogen atmosphere. The Fmoc group was removed by treatment with 25% piperidine in DMF for 20 min and 10 min followed by the previously mentioned washing steps. Afterwards the resin was treated with Fmoc-Gly-15N (1.5 equiv.), Boc-Gly-15N (1.5 equiv.) or Fmoc-Gly-1-13C (2.0 equiv.), HBTU (2.0 equiv.), HOBt (2.0 equiv.) and DIPEA (4.0 equiv.) in DMF two times for 60 min for each reactive groups. The resin was washed several times with DMF, MeOH and DCM before it was dried under vacuum.Borane reduction
A solution of borane in THF (1.0 M, 25.0 equiv. per amide) was added to the dry resin under nitrogen atmosphere in a flame-dried flask. After 30 min at ambient temperature, the reaction mixture was heated to 55 °C and kept at this temperature for two to five days depending on the length of the polyamines. Washing with THF and MeOH was followed by alkaline workup. The resin was treated with 25% piperidine in DMF (25.0 mL g−1 resin) at 55 °C for two to five days. Finally, the resin was suspended in 2% hydrazine in DMF (10 mL g−1 resin). After shaking for 30 min the resin was filtered, washed several times with DMF, MeOH, and DCM, the resin was dried under vacuum.Cleavage and isolation
The oligoamine was cleaved from the resin with 95% aqueous trifluoroacetic acid. After shaking at ambient temperature for 3 h the resin was filtered and washed with TFA. The combined filtrates were concentrated under reduced pressure and precipitated from cold, dry diethyl ether (30.0 mL). The white solid was washed three times with diethyl ether and lyophilized from water.LCPA 4
Yield 18.6 mg of yellow foam. 1H NMR (500 MHz, 300 K, DMSO-d6) δ [ppm] 9.13–8.65 (m, 6H, 1-, 5-NH2+, 8-15NH3+), 8.12–7.92 (dt, 1JH–15N −73 Hz, 2H, 8-15NH2), 3.28–3.09 (m, 4H, 7-CH2), 3.09–2.92 (m, 8H, 4-CH2, 2-CH2), 2.67–2.61 (m, 4H, 6-CH2), 2.01–1.89 (m, 4H, 3-CH2). 13C NMR (125 MHz, 300 K, DMSO-d6) δ [ppm] 44.1, 42.6, 32.7, 22.7.LCPA 6
Yield 26.5 mg of pale yellow foam. 1H NMR (500 MHz, 300 K, DMSO-d6) δ [ppm] 9.12–8.62 (m, 15H, 1-, 5-, 9-, 13-, 17-, 21-NH2+, 25-NH3+), 3.27–3.04 (m, 8H, 22-, 24-CH2), 3.04–2.80 (m, 36H, 4-, 6-, 8-, 10-, 12-, 14-, 16-, 18-, 20-CH2), 2.14–1.84 (m, 20H, 7-, 11-, 15-, 19-, 23-CH2), 1.84–1.53 (m, 4H, 2-CH2), 1.53–1.39 (m, 4H, 3-CH2). 13C NMR (125 MHz, 300 K, DMSO-d6) δ [ppm] 52.1, 44.7, 29.6, 23.1, 20.3.LCPA 8
Yield 65.4 mg of pale yellow foam. 1H NMR (500 MHz, 300 K, DMSO-d6) δ [ppm] 9.11–8.45 (m, 15H, 1-, 5-, 9-, 13-, 17-, 21-NH2+, 24-15NH3+), 3.39–3.04 (m, 12H, 4-, 22-, 23-CH2), 3.04–2.85 (m, 32H, 6-, 8-, 10-, 12-, 14-, 16-, 18-, 20-CH2), 2.11–1.87 (m, 16H, 7-, 11-, 15-, 19-CH2), 1.83–1.58 (m, 4H, 2-CH2), 1.54–1.39 (m, 4H, 3-CH2). 13C NMR (125 MHz, 300 K, DMSO-d6) δ [ppm] = 52.6, 50.1, 44.8, 29.6, 23.3, 21.0.LCPA 10
Yield 17.4 mg of pale yellow foam. 1H NMR (500 MHz, 300 K, DMSO-d6) δ [ppm] 9.14–8.44 (m, 13H, 1-, 5-, 9-, 13-, 17-, 21-NH2+, 24-NH3+), 3.30–3.05 (m, 12H, 4-CH2, 22-13CH2, 23-CH2), 3.05–2.80 (m, 32H, 6-, 8-, 10-, 12-, 14-, 16-, 18-, 20-CH2), 2.15–1.85 (m, 16H, 7-, 11-, 15-, 19-CH2), 1.76–1.59 (m, 4H, 2-CH2), 1.51–1.38 (m, 4H, 3-CH2). 13C NMR (125 MHz, 300 K, DMSO-d6) δ [ppm] 51.8, 44.2, 42.7, 29.1, 22.5, 19.9.LCPA 12
Yield 19.7 mg of pale yellow foam. 1H NMR (500 MHz, 300 K, DMSO-d6) δ [ppm] 9.09–8.52 (m, 13H, 1-, 5-, 9-, 16-NH2+, 12-15NH2+, 19-NH3+), 3.52–3.36 (m, 16H, 10-, 11-, 17-, 18-CH2), 3.29–3.09 (m, 4H, 4-CH2), 3.03–2.79 (m, 16H, 6-, 8-, 13-, 15-CH2), 2.61–2.34 (m, 8H, 7-, 14-CH2), 2.24–1.89 (m, 4H, 2-CH2), 1.79–1.59 (m, 4H, 3-CH2). 13C NMR (125 MHz, 300 K, DMSO-d6) δ [ppm] 52.2, 42.4, 34.7, 29.5, 22.7, 20.1.LCPA 14
Yield 28.3 mg of pale yellow foam. 1H NMR (500 MHz, 300 K, DMSO-d6) δ [ppm] 9.13–8.42 (m, 13H, 1-, 5-, 9-, 12-, 16-NH2+, 19-15NH3+), 3.53–3.36 (m, 16H, 10-, 11-, 17-, 18-CH2), 3.34–3.12 (m, 4H, 4-CH2), 3.02–2.76 (m, 16H, 6-, 8-, 13-, 15-CH2), 2.58–2.31 (m, 8H, 7-, 14-CH2), 2.22–1.89 (m, 4H, 2-CH2), 1.82–1.64 (m, 4H, 3-CH2). 13C NMR (125 MHz, 300 K, DMSO-d6) δ [ppm] 52.1, 42.9, 34.8, 29.2, 22.9, 20.5.Precipitation
Aqueous 29Si(OH)4 solution was prepared solving isotopically labeled Na229SiO3 (synthesized according to ref. 11 and 13) using a vortex mixer and ultrasonic bath in 2100 μL. Meanwhile 5 mg of compound 14 was dissolved in 250 μL ultrapure water. After adding 150 μL 2.4 M HCl to the 29Si(OH)4 solution and adjusting its pH value to 7 (only HCl and NaOH were used) the polyamine solution was added. The corresponding tube was then washed with 250 μL water. The washing solution which was also added to the polyamine 29Si(OH)4 solution. The pH value was then adjusted to a value of 7.00 ± 0.01 and filled up to 3 ml. The final 29Si(OH)4 concentration was 70 μM. The resulting gel was kept at room temperature for 24 h for aging, followed by centrifugation and drying of the centrifugate at 37 °C for 6 days. The dried gel was then filled in solid-state NMR rotors with a diameter of 2.5 mm.NMR
1H–13C–{29Si} CP REDOR and the implied 1H–13C CP HAHN ECHO experiments were acquired employing Rf fields of 120 kHz for 1H of 60 kHz for 13C and for 55 kHz 29Si for π and π/2 pulses. A ramp of 70–100% was applied to the proton channel while the Rf field of 13C channel kept constant at 60 kHz. The contact time was set to 4 ms and the MAS spinning rate was set to 17 kHz. For proton decoupling SPINAL64 decoupling was applied employing an Rf field of 120 kHz. 1H–29Si CP HAHN ECHO experiment was acquired employing Rf fields of 120 kHz for 1H for π/2 pulse of the CP, which also uses a ramp of 70–100%. Rf field strengths of 43.4 kHz and 55 kHz were employed for the 29Si contact pulse for the 29Si π pulses, respectively.Acknowledgements
This work was supported by the DFG Forschergruppe 2038 “Nanostructured Organic Matrices in Biological Silica Mineralization”.Notes and references
- N. Kröger, R. Deutzmann, C. Bergsdorf and M. Sumper, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 14133 CrossRef PubMed.
- M. Sumper, E. Brunner and G. Lehmann, FEBS Lett., 2005, 579, 3765 CrossRef CAS PubMed.
- S. Matsunaga, R. Sakai, M. Jimbo and H. Kamiya, ChemBioChem, 2007, 8, 1729 CrossRef CAS PubMed.
- M. C. Bridoux and A. E. Ingalls, Geochim. Cosmochim. Acta, 2010, 74, 4044 CrossRef CAS.
- T. Mizutani, H. Nagase, N. Fujiwara and H. Ogoshi, Bull. Chem. Soc. Jpn., 1998, 71, 2017 CrossRef CAS.
- M. Sumper, S. Lorenz and E. Brunner, Angew. Chem., Int. Ed., 2003, 42, 5192 CrossRef CAS PubMed.
- E. Brunner, K. Lutz and M. Sumper, Phys. Chem. Chem. Phys., 2004, 6, 854 RSC.
- P. Behrens, M. Jahns and H. Menzel, The Polyamine Silica System: A Biomimetic Model for the Biomineralization of Silica, in Handbook of Biomineralization, ed. E. Bäuerlein, Wiley-VCH, Weinheim, 2007, pp. 2–18 Search PubMed.
- D. J. Belton, S. V. Patwardhan, V. V. Annenkov, E. N. Danilovtseva and C. C. Perry, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5963 CrossRef CAS PubMed.
- V. V. Annenkov, E. N. Danilovtseva, V. A. Pal'shin, V. O. Aseyev, A. K. Petrov, A. S. Kozlov, S. V. Patwardhan and C. C. Perry, Biomacromolecules, 2011, 12, 1772 CrossRef CAS PubMed.
- K. Spinde, K. Pachis, I. Antonakaki, S. Paasch, E. Brunner and K. D. Demadis, Chem. Mater., 2011, 23, 4676 CrossRef CAS; M. Abacilar, F. Daus and A. Geyer, Beilstein J. Nanotechnol., 2015, 6, 103 CrossRef PubMed; L. Senior, M. P. Crump, C. Williams, P. J. Booth, S. Mann, A. W. Perriman and P. Cumow, J. Mater. Chem. B, 2015, 3, 2607 RSC.
- A. Jantschke, E. Koers, D. Mance, M. Weingarth, E. Brunner and M. Baldus, Angew. Chem., Int. Ed., 2015, 54, 15069 CrossRef CAS PubMed.
- D. Wisser, S. I. Brückner, F. M. Wisser, G. Althoff-Ospelt, J. Getzschmann, S. Kaskel and E. Brunner, Solid State Nucl. Magn. Reson., 2015, 66, 33 CrossRef PubMed.
- I. Ben Shir, S. Kababya, T. Amitay-Rosen, Y. S. Balazs and A. J. Schmidt, J. Phys. Chem. B, 2010, 114, 5989 CrossRef CAS PubMed.
- I. Ben Shir, S. Kababya and A. Schmidt, J. Phys. Chem. C, 2012, 116, 9691 CAS.
- T. Gullion and J. Schaefer, J. Magn. Reson., 1989, 81, 200 Search PubMed.
- A. Bernecker, R. Wieneke, R. Riedel, M. Seibt, A. Geyer and C. Steinem, J. Am. Chem. Soc., 2010, 132, 1023 CrossRef CAS PubMed.
- D. G. Hall, C. Laplante, S. Manku and J. Nagendran, J. Org. Chem., 1999, 64, 698 CrossRef CAS PubMed; F. Wang, S. Manku and D. G. Hall, Org. Lett., 2000, 2, 1581 CrossRef PubMed; S. Manku, C. Laplante, D. Kopac, T. Chan and D. G. Hall, J. Org. Chem., 2001, 66, 874 CrossRef PubMed.
- M. Bertmer and H. Eckert, Solid State Nucl. Magn. Reson., 1999, 15, 139 CrossRef CAS PubMed.
- M. Bak, J. T. Rasmussen and N. C. Nielsen, J. Magn. Reson., 2000, 147, 296 CrossRef CAS PubMed.
- M. J. Dixon, R. I. Maurer and C. Biggi, Bioorg. Med. Chem., 2005, 13, 4513 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR, HR-MS and HPLC spectra for all LCPAs and peptide precursors together with 1H NMR and HPLC spectra for the peptides. See DOI: 10.1039/c6ra19624a |
| This journal is © The Royal Society of Chemistry 2016 |