
Application of a tandem air-agitated liquid–liquid microextraction technique based on solidification of floating organic droplets as an efficient extraction method for determination of cholesterol-lowering drugs in complicated matrices†
Abstract
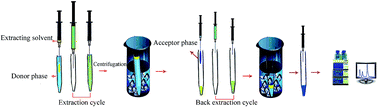
A simple and sensitive extraction method termed tandem air-agitated liquid–liquid microextraction based on solidification of floating organic droplets (TAALLME-SFO) is introduced. Based on this method, the three cholesterol-lowering drugs rosuvastatin, atorvastatin, and gemfibrozil, as the model analytes, were first extracted from a relatively large volume (10 mL) of an acidic donor phase into a small volume (105 μL) of an intermediate phase (a solidifiable organic solvent), and were then simply back-extracted into a smaller volume of a basic acceptor phase (55 μL) by micro-air-agitated liquid–liquid microextraction based on solidification of floating organic droplets (μ-AALLME-SFO). By performing this convenient extraction method, a high sample clean-up was obtained. The response surface methodology (RSM) combined with the desirability function (DF) was applied to the optimization of the effective parameters involved to reach the maximum extraction efficiency of the drugs. The pH values of the donor and acceptor phases and the volumes of the organic solvent (μL) and acceptor phase (μL) were obtained to be 3.0, 12, 105, and 55, respectively. Under the optimal experimental conditions, the TAALLME-SFO-HPLC-UV method provided good linearity for all the target analytes in the range of 2–3000 ng mL−1 with coefficients of determination (R2) ≥ 0.995, and the intra-day and inter-day precisions varied in the ranges of 4.2–5.3 and 5.7–6.6, respectively. The limits of detection (LODs) were in the range of 0.5–1.0 ng mL−1. Consequently, this new microextraction technique was demonstrated to be suitable for the extraction and determination of the studied drugs in human plasma and wastewater samples.
 Please wait while we load your content...
Please wait while we load your content...

