
Solution and solid-state fluorescence of 2-(2′-hydroxyphenyl)-1,5-benzodiazepin-2-one (HBD) borate complexes†
Abstract
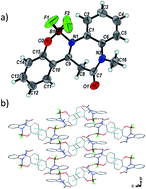
A new family of fluorescent 1,5-benzodiazepin-2-one (HBD) borate complexes was prepared in good yields, and fully characterized by means of MS, NMR and IR spectroscopy, as well as X-ray crystal structure analysis for compound 13. Unlike their uncomplexed congeners, most of these cyclic boranils were emissive both in solution and the solid state, with maxima in the range of 426–596 nm. A systematic study of substituent effects revealed that the presence of a halogen atom specifically at position 8 of the fused-aromatic ring system led to an increase in fluorescence intensity in solution while electron rich substituents tended to extinguish the photoluminescence. Finally, a proof-of-concept study highlighted that the amide moiety of the benzodiazepinone framework could be functionalized with a chemical handle useful for subsequent specific modifications.
 Please wait while we load your content...
Please wait while we load your content...

