
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of dimeric analogs of adenophostin A that potently evoke Ca2+ release through IP3 receptors†
Amol M.
Vibhute
a,
Poornenth
Pushpanandan
a,
Maria
Varghese
a,
Vera
Koniecnzy
b,
Colin W.
Taylor
b and
Kana M.
Sureshan
*a
aSchool of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram, Kerala 695016, India. E-mail: kms@iisertvm.ac.in; Web: http://kms514.wix.com/kmsgroup
bDepartment of Pharmacology, University of Cambridge, Tennis Court Road, Cambridge, CB2 1PD, UK
First published on 5th September 2016
Abstract
Inositol 1,4,5-trisphosphate receptors (IP3Rs) are tetrameric intracellular channels through which many extracellular stimuli initiate the Ca2+ signals that regulate diverse cellular responses. There is considerable interest in developing novel ligands of IP3R. Adenophostin A (AdA) is a potent agonist of IP3R and since some dimeric analogs of IP3R ligands are more potent than the corresponding monomer; we considered whether dimeric AdA analogs might provide agonists with increased potency. We previously synthesized traizolophostin, in which a simple triazole replaced the adenine of AdA, and showed it to be equipotent to AdA. Here, we used click chemistry to synthesize four homodimeric analogs of triazolophostin, connected by oligoethylene glycol chains of different lengths. We evaluated the potency of these analogs to release Ca2+ through type 1 IP3R and established that the newly synthesized dimers are equipotent to AdA and triazolophostin.
Introduction
Inositol 1,4,5-trisphosphate (IP3, 1, Fig. 1) is an important secondary messenger that evokes Ca2+ release from intracellular stores through its interaction with IP3 receptors (IP3R) in the endoplasmic reticulum.1 IP3R are large tetrameric proteins, within which IP3 binding to each of the four subunits is required to initiate opening of the Ca2+-permeable channel.2 High-resolution structures of the IP3-binding core (IBC, residues 224–604) have defined the interactions of IP3 with IP3R.3 More recently, structures of the N-terminal region (residues 1–604)4 alongside a structure of the complete IP3R derived from cryo-electron microscopy have begun to suggest how IP3 binding might trigger the opening of the intrinsic pore of IP3R.5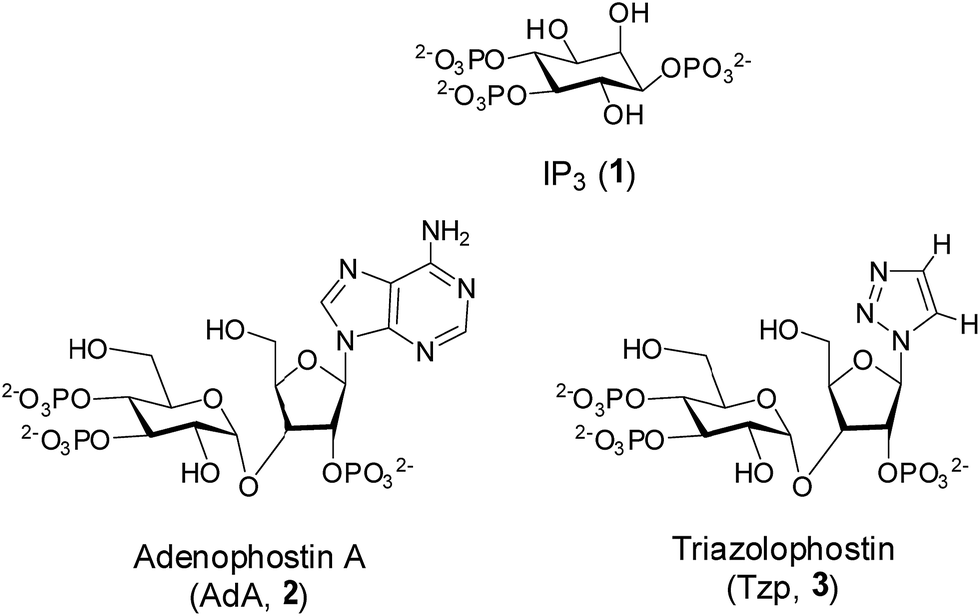 | ||
| Fig. 1 The structures of IP3 (1), adenophostin A (2) and triazolophostin (3). | ||
There is continuing interest in the development of potent agonists and antagonists of IP3R.6 The fungal metabolite, adenophostin A (AdA, 2, Fig. 1), binds to IP3R with greater affinity than IP3 and it is more potent than IP3 in evoking Ca2+ release.7 AdA analogs with a nucleobase or base-surrogate are also more potent than IP3.8 Molecular docking8j,m,9 and mutation studies10 suggest that a cation–π interaction between the adenine moiety of AdA and Arg504 within the IBC contributes to the increased affinity of AdA. We recently reported synthesis of a library of active AdA analogs, triazolophostins, by using a click chemistry approach.11 These potent analogs have substituted triazoles as adenine surrogates. The simplest analog, triazolophostin (3, Fig. 1) was equipotent with AdA.
Multimeric ligands often have greater affinity than monomeric ligands.12 This can be due to simultaneous binding to more than one binding site or a statistical effect arising from the local increase in ligand concentration.13 The former is unlikely for IP3R because the orientation of the IP3-binding sites within the tetrameric IP3R is unlikely to allow simultaneous binding of two ligands linked by a short tether.4b,14
A few multimeric ligands of IP3R have been reported. Before the location of the IP3-binding sites within IP3R was known, clustered bi- and tetra-dentate analogs of ribophostin (4, Fig. 2A) were synthesized, anticipating that if the spacing between the linked ligands was appropriate they might bind simultaneously to the four IP3-binding sites.15 However, the potencies of the monomeric and polymeric ligands were rather similar. Several homodimeric16 and heterodimeric17 ligands of IP3 (5–10, Fig. 2B), particularly those with short linkers, were shown to bind to IP3R with increased affinity.13d Very recently, dimers of 2-O-Bt-IP4/IP5 (11, Fig. 2C) were shown to be antagonists of IP3Rs.18 These results demonstrate that dimeric IP3R ligands can provide useful tools, some of which have greater affinity than the monomeric ligands. We therefore considered whether dimers of AdA might be more potent than AdA.
 | ||
| Fig. 2 The representative structures of (A) ribophostin dimer 4, (B) homo and hetero dimers of IP3 (5–10) and (C) dimers of 2-Bt-IP4/IP511. | ||
Results and discussion
As the synthesis of AdA dimers is challenging, we decided to make oligoethylene glycol-tethered dimers of triazolophostin (Fig. 3). We envisaged that use of click reaction19 with a linker connected to alkyne at both termini would ensure both formation of triazole and link the two monomers in one step. Previous studies suggested that short linkers were most likely to improve the affinity of homodimers.13d We therefore selected spacers smaller than hexaethylene glycol. The linkers 14a–d were synthesized by slightly modifying previously reported procedures.20 The oligoethylene glycols were first co-evaporated with toluene and then treated with sodium hydride in the presence of excess propargyl bromide to get dipropargyl polyethylene glycols 14a–d in good to excellent yields. The azide 13 was synthesized from glucose and xylose by several protection–deprotection reactions followed by phosphorylation as reported earlier.11 The azide 13 was then treated with dialkynyl polyethylene glycols 14a–d in the presence of Cu(I) catalyst to get fully protected triazolophostin dimers 15a–d in good yields. The debenzylation of protected triazolophostin dimers 15a–d was carried out using transfer hydrogenolysis in the presence of palladium and cyclohexene under reflux condition and the products were purified by ion-exchange chromatography to yield dimers 12a–d, in excellent yields (Scheme 1). | ||
| Fig. 3 The structure of dimeric analogs of triazolophostin 12. | ||
 | ||
Scheme 1 Synthesis of triazolophostin dimers. Reagents and conditions: (a) ref. 11; (b) Cu, CuSO4, H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :tBuOH (1:1, v/v), rt, 24 h; (c) Pd(OH)2/C, cyclohexene, MeOH:H2O (10:1, v/v), 80 °C. 4 h; (a), n = 2; (b), n = 3; (c), n = 4; (d), n = 6. :tBuOH (1:1, v/v), rt, 24 h; (c) Pd(OH)2/C, cyclohexene, MeOH:H2O (10:1, v/v), 80 °C. 4 h; (a), n = 2; (b), n = 3; (c), n = 4; (d), n = 6. | ||
The dimeric ligands 12a–d were screened for their abilities to evoke Ca2+ release through IP3R (Table 1, Fig. 4). All four dimers were full agonists of IP3R, more potent than IP3, but similar in their potency to AdA and the monomer, triazolophostin. The similar potencies of 12a–d irrespective of their tether length suggest that these ligands might be interacting with IP3R1 in monodentate fashion.
| Ligand | pEC50 | EC50 (nM) | EC50 w.r.t. 1b | Max. response (%) | n H |
|---|---|---|---|---|---|
|
a Maximal Ca2+ release, the half-maximally effective ligand concentration (EC50), −logEC50 (pEC50) and Hill coefficient (nH) are shown as means ± SEM (n = 3).
b The EC50 value of each ligand is also shown relative to that for IP3 (1) (EC150/ECanalog50).
|
|||||
| IP3 (1) | 6.72 ± 0.12 | 190.5 | 1 | 69 ± 3 | 1.40 ± 0.16 |
| Monomer (3) | 7.86 ± 0.17 | 13.8 | 13.8 | 65 ± 1 | 1.66 ± 0.21 |
| 12a | 7.83 ± 0.18 | 14.8 | 12.9 | 68 ± 2 | 1.33 ± 0.12 |
| 12b | 7.85 ± 0.13 | 14.1 | 13.5 | 66 ± 1 | 1.89 ± 0.13 |
| 12c | 7.62 ± 0.11 | 24.0 | 7.9 | 61 ± 3 | 1.60 ± 0.16 |
| 12d | 7.84 ± 0.12 | 14.4 | 13.2 | 60 ± 1 | 1.94 ± 0.47 |
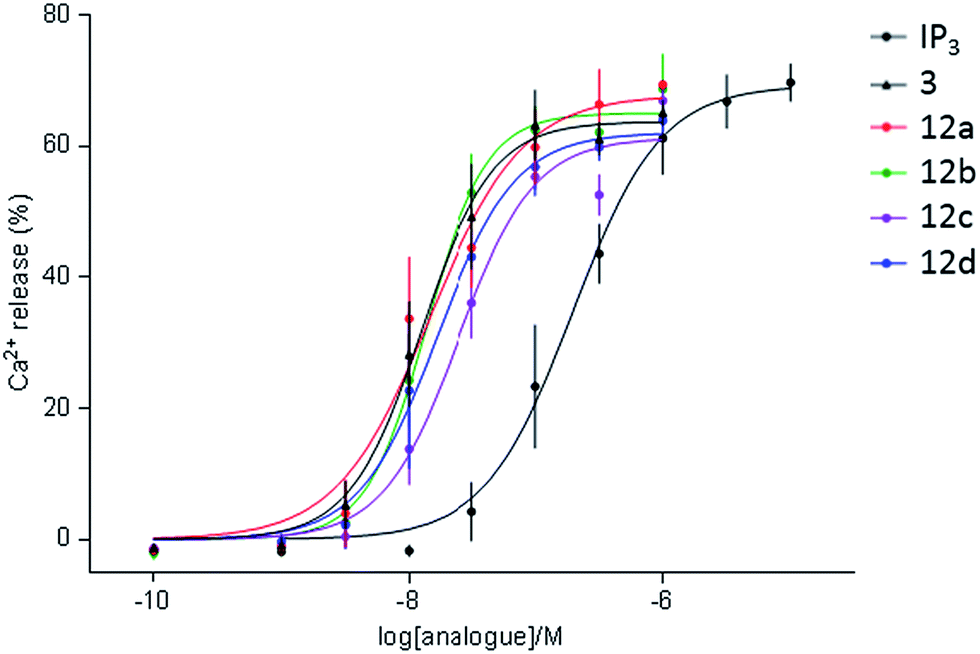 | ||
| Fig. 4 Summary of Ca2+ release from permeabilized DT40-IP3R1 cells evoked by IP3, monomer 3 and its dimeric analogs 12a–d. | ||
Conclusions
In conclusion, based on several previous reports that dimeric IP3R ligands can be more potent than the corresponding monomers, we anticipated that dimers of AdA might have increased potency. We used click chemistry to synthesize dimers of a potent analog of AdA (triazolophostin) linked by spacers of different length. In assays of Ca2+ release through IP3R, the dimeric ligands were no more potent than the corresponding monomer (3). This suggests that whereas dimeric derivatives of IP3 have reduced efficacy but improved affinity,10,21 dimerization of AdA analogs does not improve their affinity.Experimental section
General methods
The chemicals were purchased from commercial sources and used as received. The TLC plates were visualized under UV light and by dipping plates into either phosphomolybdic acid in MeOH or sulphuric acid in ethanol, followed by heating. All NMR experiments were carried out on a 500 MHz NMR spectrometer and at room temperature. Tetramethylsilane (TMS, δ 0.0 ppm) or the solvent reference (CDCl3, δ 7.26 ppm; D2O, δ 4.79 ppm) relative to TMS were used as the internal standard. The data are reported as follows: chemical shift in ppm (δ) (multiplicity [singlet (s), doublet (d), doublet of doublet (dd), triplet (t), quartet (q), and multiplet (m)], coupling constants [Hz], integration and peak identification). All NMR signals were assigned on the basis of 1H NMR, 13C NMR, COSY and HMQC experiments. 13C NMR spectra were recorded with complete proton decoupling. Carbon chemical shifts are reported in ppm (δ) relative to TMS with the respective solvent resonance as the internal standard. The concentration of the compounds for 1H NMR was 5 mg per 0.5 mL and for 13C NMR it was 20 mg per 0.5 mL for protected compounds and 5–7 mg per 0.5 mL for final compounds in case of 1H and 13C NMR. Modified Brigg's phosphate assay22 was employed to quantify each triazolophostin 12a–d. Silica gel 230–400 mesh was used to perform flash column chromatography.General procedure for syntheses of fully protected triazolophostin dimers
To a solution of azide 13 (0.144 mmol) and dialkynyl PEG 14a–d (0.072) in H2O/tBuOH (1/1, v/v, 2 mL) was added Cu (0.036 g, 0.57 mmol) and CuSO4 (8 mg, 0.028 mmol) and stirred at room temperature for 24 h. The reaction was monitored by TLC. When the TLC showed complete disappearance of the azide 13, the mixture was filtered through a Celite bed and was partitioned between ethyl acetate and water. The organic layer was washed with brine. The organic layer was dried over anhyd. sodium sulphate, filtered and concentrated under reduced pressure. The residue thus obtained was purified by flash column chromatography using a mixture of acetone, diethyl ether and petroleum ether (4:2:15 v/v/v) as eluent to get pure 15a–d as a colourless gum.
General procedure for syntheses of triazolophostin dimers 12a–d
The protected triazolophostin dimers 15a–d (0.15–0.175 g, 0.05–0.055 mmol) were treated with cyclohexene (3 mL) and Pd(OH)2 (20% on carbon, 50 mg) in a mixture of methanol and water (9:1 v/v, 10 mL) at 80 °C for 4 h. The reaction mixture was then cooled, filtered through a membrane filter, washed successively with methanol and water. The combined filtrate was evaporated under reduced pressure. The crude product thus obtained was purified by ion-exchange column chromatography on Q-Sepharose matrix using 0–1.0 M TEAB as eluent to get pure triazolophostin dimers 12a–d.
Biological assay
Ca2+ release from the intracellular stores of saponin-permeabilized DT40 cells expressing only type 1 IP3Rs was measured using a low-affinity Ca2+ indicator (Mag-fluo-4) trapped within the endoplasmic reticulum as described previously.11 Briefly, Ca2+ uptake was initiated by addition of 1.5 mM MgATP in cytosol-like medium (140 mM KCl, 20 mM NaCl, 1 mM EGTA, 20 mM PIPES, pH 7.0, free [Ca2+] ∼220 nM after addition of ATP) containing p-trifluoromethoxyphenylhydrazone (FCCP) to inhibit mitochondria. After about 120 s, the triazolophostin analogs were added with cyclopiazonic acid (10 μM) to inhibit further Ca2+ uptake. Ca2+ release was assessed 10–20 s after addition of the analog, and expressed as a fraction of the ATP-dependent Ca2+ uptake.Acknowledgements
AMV thanks the University Grants Commission (UGC) India for a Senior Research Fellowship (SRF) during this work. KMS thanks Department of Science and Technology (DST) India for Swarnajayanti Fellowship, Ramanujan Fellowship and for financial support. CWT and VK were supported by the Wellcome Trust.Notes and references
- (a) M. J. Berridge, Nature, 1993, 361, 315 CrossRef CAS PubMed; (b) B. V. L. Potter and D. Lampe, Angew. Chem., Int. Ed. Engl., 1995, 34, 1933 CrossRef CAS; (c) J. K. Foskett, C. White, K. H. Cheung and D. O. Mak, Phys. Rev., 2007, 87, 593 CAS; (d) M. J. Berridge, Biochim. Biophys. Acta, 2009, 1793, 933 CrossRef CAS PubMed; (e) C. W. Taylor and S. C. Tovey, Cold Spring Harbor Perspect. Biol., 2010, 2, a004010 CAS.
- K. J. Alzayady, L. Wang, R. Chandrasekhar, L. E. Wagner II, F. Van Petegem and D. I. Yule, Sci. Signaling, 2016, 9, ra35 CrossRef PubMed.
- I. Bosanac, J.-R. Alattia, T. K. Mal, J. Chan, S. Talarico, F. K. Tong, K. I. Tong, F. Yoshikawa, T. Furuichi, M. Iwai, T. Michikawa, K. Mikoshiba and M. Ikura, Nature, 2002, 420, 696 CrossRef CAS PubMed.
- (a) C. C. Lin, K. Baek and Z. Lu, Nat. Struct. Mol. Biol., 2011, 18, 1172 CrossRef CAS PubMed; (b) M.-D. Seo, S. Velamakanni, N. Ishiyama, P. B. Stathopulos, A. M. Rossi, S. A. Khan, P. Dale, C. Li, J. B. Ames, M. Ikura and C. W. Taylor, Nature, 2012, 483, 108 CrossRef CAS PubMed.
- G. Fan, M. L. Baker, Z. Wang, M. R. Baker, P. A. Sinyagovskiy, W. Chiu, S. J. Ludtke and I. I. Serysheva, Nature, 2015, 527, 336 CrossRef CAS PubMed.
- (a) H. Saleem, S. C. Tovey, T. Rahman, A. M. Riley, B. V. L. Potter and C. W. Taylor, PLoS One, 2012, 8, e54877 Search PubMed; (b) H. Saleem, S. C. Tovey, T. F. Molinski and C. W. Taylor, Br. J. Pharmacol., 2014, 171, 3298 CrossRef CAS PubMed.
- (a) M. Takahashi, T. Kagasaki, T. Hosoya and S. Takahashi, J. Antibiot., 1993, 46, 1643 CrossRef CAS PubMed; (b) J. Hirota, T. Michikawa, A. Miyawaki, M. Takahashi, K. Tanzawa, I. Okura and K. Mikoshiba, FEBS Lett., 1995, 368, 248 CrossRef CAS PubMed.
- (a) J. S. Marchant, M. D. Beecroft, A. M. Riley, D. J. Jenkins, R. D. Marwood, C. W. Taylor and B. V. L. Potter, Biochemistry, 1997, 36, 12780 CrossRef CAS PubMed; (b) S. Shuto, K. Tatani, Y. Ueno and A. Matsuda, J. Org. Chem., 1998, 63, 8815 CrossRef CAS; (c) R. D. Marwood, A. M. Riley, V. Correa, C. W. Taylor and B. V. L. Potter, Bioorg. Med. Chem. Lett., 1999, 9, 453 CrossRef CAS PubMed; (d) H. Hotoda, K. Murayama, S. Miyamoto, Y. Iwata, M. Takahashi, Y. Kawase, K. Tanzawa and M. Kaneko, Biochemistry, 1999, 38, 9234 CrossRef CAS PubMed; (e) M. Kashiwayanagi, K. Tatani, S. Shuto and A. Matsuda, Eur. J. Neurosci., 2000, 12, 606 CrossRef CAS PubMed; (f) S. Shuto, M. Terauchi, Y. Yahiro, H. Abe, S. Ichikawa and A. Matsuda, Tetrahedron Lett., 2000, 41, 4151 CrossRef CAS; (g) F. Chretien, N. Moitessier, F. Roussel, J.-P. Mauger and Y. Chapleur, Curr. Org. Chem., 2000, 4, 513 CrossRef CAS; (h) H. Abe, S. Shuto and A. Matsuda, J. Org. Chem., 2000, 65, 4315 CrossRef CAS PubMed; (i) V. Correa, A. M. Riley, S. Shuto, G. Horne, E. P. Neruo, R. D. Marwood, B. V. L. Potter and C. W. Taylor, Mol. Pharmacol., 2001, 59, 1206 CAS; (j) H. J. Rosenberg, A. M. Riley, A. J. Laude, C. W. Taylor and B. V. L. Potter, J. Med. Chem., 2003, 46, 4860 CrossRef CAS PubMed; (k) C. N. Borissow, S. J. Black, M. Paul, S. C. Tovey, S. G. Dedos, C. W. Taylor and B. V. L. Potter, Org. Biomol. Chem., 2005, 3, 245 RSC; (l) K. M. Sureshan, M. Trusselle, S. C. Tovey, C. W. Taylor and B. V. L. Potter, Chem. Commun., 2006, 2015 RSC; (m) T. Mochizuki, Y. Kondo, H. Abe, S. C. Tovey, S. G. Dedos, C. W. Taylor, M. Paul, B. V. L. Potter, A. Matsuda and S. Shuto, J. Med. Chem., 2006, 49, 5750 CrossRef CAS PubMed; (n) A. M. Rossi, A. M. Riley, B. V. L. Potter and C. W. Taylor, Curr. Top. Membr., 2010, 66, 209 CAS; (o) K. M. Sureshan, A. M. Riley, M. P. Thomas, S. C. Tovey, C. W. Taylor and B. V. L. Potter, J. Med. Chem., 2012, 55, 1706 CrossRef CAS PubMed; (p) H. Saleem, S. C. Tovey, A. M. Riley, B. V. L. Potter and C. W. Taylor, PLoS One, 2013, 8, e58027 CAS.
- K. M. Sureshan, M. Trusselle, S. C. Tovey, C. W. Taylor and B. V. L. Potter, J. Org. Chem., 2008, 73, 1682 CrossRef CAS PubMed.
- A. M. Rossi, K. M. Sureshan, A. M. Riley, B. V. L. Potter and C. W. Taylor, Br. J. Pharmacol., 2010, 161, 1070 CrossRef CAS PubMed.
- A. M. Vibhute, V. Konieczny, C. W. Taylor and K. M. Sureshan, Org. Biomol. Chem., 2015, 13, 6698 CAS.
- (a) R. H. Kramer and J. W. Karpen, Nature, 1998, 395, 710 CrossRef CAS PubMed; (b) M. Mammen, S.-K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754 CrossRef; (c) P. S. Portoghese, J. Med. Chem., 2001, 44, 2259 CrossRef CAS PubMed.
- (a) N. L. Pohl and L. L. Kiessling, Synthesis, 1999, 1515 CrossRef CAS; (b) L. L. Kiessling, J. E. Gestwicki and L. E. Strong, Curr. Opin. Chem. Biol., 2000, 4, 696 CrossRef CAS PubMed; (c) J. E. Gestwicki, C. W. Cairo, L. E. Strong, K. A. Oetjen and L. L. Kiessling, J. Am. Chem. Soc., 2002, 124, 14922 CrossRef CAS PubMed; (d) A. M. Riley, A. J. Laude, C. W. Taylor and B. V. L. Potter, Bioconjugate Chem., 2004, 15, 278 CrossRef CAS PubMed.
- S. J. Ludtke, T. P. Tran, Q. T. Ngo, V. Y. Moiseenkova-Bell, W. Chiu and I. I. Serysheva, Structure, 2011, 19, 1192 CrossRef CAS PubMed.
- (a) M. de Kort, A. R. P. M. Valentijn, G. A. van der Marel and J. H. van Boom, Tetrahedron Lett., 1997, 38, 7629 CrossRef CAS; (b) M. de Kort, V. Correa, A. R. P. M. Valentijn, G. A. van der Marel, B. V. L. Potter, C. W. Taylor and J. H. van Boom, J. Med. Chem., 2000, 43, 3295 CrossRef CAS PubMed.
- A. M. Riley and B. V. L. Potter, Chem. Commun., 2000, 983 RSC.
- A. M. Rossi, A. M. Riley, S. C. Tovey, T. Rahman, O. Dellis, E. J. A. Taylor, V. G. Veresov, B. V. L. Potter and C. W. Taylor, Nat. Chem. Biol., 2009, 5, 631 CrossRef CAS PubMed.
- V. Konieczny, J. G. Stefanakis, E. D. Sitsanidis, N.-A. T. Ioannidou, N. V. Papadopoulos, K. C. Fylaktakidou, C. W. Taylor and A. E. Koumbis, Org. Biomol. Chem., 2016, 14, 2504 CAS.
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS; (b) J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249 RSC.
- (a) Z.-J. Yao, H.-P. Wu and Y.-L. Wu, J. Med. Chem., 2000, 43, 2484 CrossRef CAS PubMed; (b) K. Tanaka, H. Sagae, K. Toyoda and K. Noguchi, Eur. J. Org. Chem., 2006, 3575 CrossRef CAS.
- (a) L. Glouchankova, U. M. Krishna, B. V. L. Potter, J. R. Falck and I. Bezprozvanny, Mol. Cell Biol. Res. Commun., 2000, 3, 153 CrossRef CAS PubMed; (b) S. A. Morris, E. P. Nerou, A. M. Riley, B. V. L. Potter and C. W. Taylor, Biochem. J., 2002, 367, 113 CrossRef CAS PubMed.
- D. Lampe, C. Liu and B. V. L. Potter, J. Med. Chem., 1994, 37, 907 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectral data for all the new compounds. See DOI: 10.1039/c6ra19413c |
| This journal is © The Royal Society of Chemistry 2016 |