
Ultrasmall polymersomes of poly-α,β-(N-2-hydroxyethyl L-aspartamide)-graft-poly(L-lactic acid) copolymers as a potential drug carrier†
Hyun Jin Leea,
Seung-Hae Kwonb and
Kwang-Suk Jang*c
aDepartment of Chemical & Biomolecular Engineering, Korea Advanced Institute of Science and Technology, Daejeon 34141, Republic of Korea
bDivision of Bio-imaging, Korea Basic Science Institute, Chun-Cheon 24341, Republic of Korea
cDepartment of Chemical Engineering and Research Center of Chemical Technology, Hankyong National University, Anseong 17579, Republic of Korea. E-mail: jang@hknu.ac.kr
First published on 5th September 2016
Abstract
Ultrasmall polymersomes of poly-α,β-(N-2-hydroxyethyl L-aspartamide)-graft-poly(L-lactic acid), 40–45 nm in diameter, were prepared based on a well-defined graft copolymer architecture and well-managed self-assembly process. The ultrasmall polymersomes are a proposed drug delivery platform based on their suitable size, narrow size distribution, uniform morphology, and high thermodynamic stability, which satisfy prerequisites for systemic drug delivery carriers. The functionality and versatility of this system as a potential general drug delivery platform are supported by its biodegradability, favorable modifiable chemical formula, and flexible polymersome size that could be tuned to a nanoscopic range by manipulating the thermodynamic balance of the self-assembly process.
1. Introduction
In selective solvents, amphiphilic copolymers self-assemble into colloidal nanostructures such as micelles, cylinders, lamella, and vesicles according to the hydrophilic/hydrophobic ratio of copolymer molecules.1–4 The aqueous self-assembly of amphiphilic copolymers to fabricate nanoscale self-aggregates of various morphologies has drawn considerable attention owing to the unique physicochemical characteristics. The polymeric self-aggregates have been used in diverse applications based on their well-defined structures consisting of hydrophilic and hydrophobic compartments and their miscellaneous functionalities, facilitated by versatile polymer chemistry. One topical application is in the development of drug delivery systems for cancer treatment owing to their high capacity for drug payload, controlled drug release, and prolonged blood circulation of the polymeric self-aggregates as drug carriers.5–7In particular, the polymer vesicles, which are also known as polymersomes, have been at the core of this research trend over the past decade.8–14 Polymersomes prepared from amphiphilic copolymers with a large hydrophobic content show a liposome-like structure composed of bilayered polymer membranes enclosing a fluid-filled hydrophilic cavity. Despite their structural resemblance to liposomes, polymersomes show a higher stability and stronger functionality owing to their advantageous polymeric nature such as the strong interaction between the long polymer chains and considerable chemical versatility. The thick hydrophobic core of the polymer bilayer can be loaded with hydrophobic substances.11 The inner hydrophilic compartment that is protected by polymer membranes can be used to entrap various hydrophilic bioactive agents with different charges, size, and shapes such as single small molecule drugs, peptides, proteins, and oligonucleotides.12–14
Although polymersomes are promising because of their ability to entrap various biofunctional molecules with different solubility and molecular size, they need improvements in terms of size, size dispersity, and morphological uniformity. There have been frequent reports of the formation of polymersomes with a broad size distribution within several hundreds of nanometers, which is out of the recommended range for achieveing prolonged circulation time and an enhanced permeation and retention (EPR) effect.15 In addition, the lack of morphological uniformity such as the existence of both unilamellar and multilamellar vesicles and a mixture of different shapes including spheres, oblate forms, and tubules have often been observed. These are critical characteristics that would the performance of polymersomes as drug carriers because size and shape are influencing factors not only for drug loading and release, but also for blood circulation time, whole body distribution, and intracellular uptake and distribution.16–19 Therefore, the development of polymersomes with a small particle size <200 nm, narrow size distribution, and well-defined uniform morphology along with desirable material properties is highly desired and demanded.
Here, we demonstrate ultrasmall polymersomes with a diameter <50 nm prepared from a biodegradable graft copolymer, poly-α,β-(N-2-hydroxyethyl L-aspartamide)-graft-poly(L-lactic acid) (PHEA-g-PLA), as a potential general drug delivery platform. This is the continuation of a previous study, which urgently reported the first polymersomes from the graft copolymer system with emphasis on the novel graft copolymer, PHEA-g-PLA, and its self-assembling behavior forming micelles and vesicles as a function of PLA content.20 It was presumed that the strong intra- and inter-molecular entanglement of graft copolymers would hinder vesicle formation and would rather induce micelle-like aggregates. However, properly designed graft copolymers can self-assemble into a well-defined vesicular structure. The interesting molecular architecture, well-organized self-aggregate structure, and the biodegradability of PHEA-g-PLA copolymers have encouraged us to pursue further investigation.
In the present study, a thorough physicochemical examination was first performed to both quantitatively and qualitatively analyze the micelles and ultrasmall vesicles to demonstrate that both self-aggregates are suitable candidates for systemic drug delivery carriers. Then, focusing on hydrophilic molecule delivery as distinguishing characteristic of the vesicles, we show that the ultrasmall polymersomes can entrap and deliver the water-soluble anticancer drug, doxorubicin hydrochloride (DOX). Moreover, the modification of polymersomes with the cyclic arginylglycylaspartic acid (Arg-Gly-Asp, RGD) peptide as an active cancer-targeting ligand was demonstrated. Polymersomes with a very small size similar to those in this study have been rarely reported.21 The present study show the first example of a cyclic RGD peptide-functionalized polymersome. In addition, the alteration of the vesicle size to the desirable nanoscopic range of <200 nm using a minor modification of the preparation method is another versatile feature of the present system.
In this study, ultrasmall polymersomes with a diameter of 40 nm that have not been previously reported in other studies were prepared. The geometrical factor of nonlinear graft copolymer and its effect on self-assembling behavior and resultant self-aggregate structure are thoroughly discussed. The polymersomes showed a unilamellar spherical vesicular morphology with a narrow size distribution while no other structures or morphologies such as micelles, cylinders, as well as ellipsoidal, tubular, and multilamellar vesicles were present. This highly defined uniform morphology is very advantageous as a potential general drug delivery platform based on a polymersome carrier. Furthermore, the other advantages of the present systems include a critical aggregation concentration as low as several nanomolar, which suggest the high thermodynamic stability of the polymersome carrier. In addition, PLA is a hydrolytic degradable aliphatic polyester while PHEA is a water-soluble synthetic poly(amino acid) and, therefore, the resultant in vivo-degradable and absorbable system is an excellent alternative to poly(ethylene glycol). The PHEA-g-PLA copolymer platform is favorable to upgrading to a multivalent or multifunctional system using abundant hydroxyl and residual amino groups similar to what we achieved by functionalization with the cyclic RGD peptide.
2. Experimental section
2.1. Synthesis of polymers
The PLA was synthetized as described previously.20 Briefly, PLA was synthesized by ring-opening polymerization of L-lactide using 1-octanol as the initiator and stannous octoate as the catalyst. L-Lactide was purified by recrystallization from ethyl acetate. Distilled chloroform was used as a solvent for the polymerization reaction. After polymerization at 130 °C for 10 h under N2, the PLA was purified by precipitation in ether. The successful synthesis of the PLA was confirmed by determining its characteristics such as the molecular weight (MW) using proton nuclear magnetic resonance (1H-NMR) with a Bruker AMX 500 instrument. The MW of PLA was determined using matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectroscopy (Voyager DE-STR from Applied Biosystems Inc.) with α-cyano-4-hydroxycinnamic acid as matrix and reflector mode as the detection condition. The hydroxyl group of the PLA chain terminus was activated with a coupling agent, carbonyldiimidazole (CDI) to form PLA-CDI for the grafting reaction.The PHEA-g-PLA was synthesized as reported previously.20 Briefly, the precursor polymer, poly(succinimide) (PSI), was pre-synthesized by the acid-catalyzed polycondensation of L-aspartic acid using phosphoric acid as the catalyst at 180 °C under N2. The backbone PHEA polymer containing primary amine groups (PHEA-NH2) was prepared by reacting PSI with a 0.9 equivalent of ethanolamine for 6 h, followed by an excess amount of ethylenediamine for 6 h at 40 °C in dry dimethylformamide (DMF). The PHEA-NH2 was collected by precipitation with ether and analyzed using 1H-NMR. The PLA was grafted onto the backbone polymer through a carbamate bond by a reaction between the amine groups in PHEA-NH2 and the CDI-activated PLA chain terminus. The grafting reaction was conducted in dry DMF at 70 °C for 12 h. The reaction mixture was purified by extensive dialysis against DMF, followed by distilled deionized (DDI) water. After freeze-drying the solution, the final PHEA-g-PLA graft copolymer product was obtained. Five PHEA-g-PLA copolymers with different grafting degrees of PLA were prepared. The molecular compositions of the PHEA-g-PLA copolymers were verified using 1H-NMR and elemental analysis (Eager 200, CE Instruments).
Cyclic RGD peptide with a sequence of c(RGDfC) (cyclic Arg-Gly-Asp-Phe-Cys) was purchased from Peptron (Daejeon, Republic of Korea). The peptide was conjugated to PHEA-g-PLA using a coupling agent, N-hydroxysuccinimide-(ethylene oxide)2-maleimide (NHS-EO2-maleimide, from Pierce) to yield PHEA-g-PLA–c(RGDfC). The PHEA-g-PLA was first reacted with NHS-EO2-maleimide in dry DMF at room temperature for 6 h and then purified by dialysis against DDI water. The conjugation reaction of c(RGDfC) was conducted in a mixed solvent of dimethyl sulfoxide (DMSO) and DDI water (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30, v/v) at room temperature for 6 h. The PHEA-g-PLA–c(RGDfC) was purified by dialysis against DDI water, and the conjugation was verified by 1H-NMR.
:30, v/v) at room temperature for 6 h. The PHEA-g-PLA–c(RGDfC) was purified by dialysis against DDI water, and the conjugation was verified by 1H-NMR.
2.2. Preparation and characterization of self-aggregates
The self-aggregates were prepared using the following precipitation–dialysis method.20 Briefly, 10 mg of the PHEA-g-PLA copolymer was dissolved in 1 mL DMSO, which is a good solvent for both PHEA and PLA. The polymer solution in DMSO was added dropwise to 10 mL DDI water while the solution was vigorously stirred. The mixed solution was then dialyzed against DDI water, and the aqueous solution of the self-aggregates was filtered through a 0.45 μm polyvinylidene fluoride (PVDF) filter to remove any dust particles.The CAC values were determined from the fluorescence excitation spectra of pyrene, which was added to the PHEA-g-PLA copolymer solutions of various concentrations. The pyrene concentration in the polymer solution was adjusted to 6.0 × 10−7 M. The emission monochromator was set at 390 nm and the excitation and emission slit widths were 2.5 and 5.0 nm, respectively. The change in the ratio between the intensities at 333 and 336 nm (intensity ratio, I333/I336) was used as an indicator of the self-aggregate formation. To measure the size of the self-aggregates, dynamic light scattering (DLS) measurement was performed using an apparatus (Brookhaven Instruments Inc.) equipped with a 673 nm He–Ne laser at a scattering angle of 90°. The size distribution of the self-aggregates and the mean hydrodynamic diameter were derived from a non-negative least squares (NNLS) algorithm. The self-aggregates were visualized using transmission electron microscopy (TEM) with an LEO-912AB OMEGA instrument (Carl Zeiss, Inc.). The self-aggregates in aqueous solution were negatively stained with 2 wt% phosphotungstic acid (PTA) for contrast adjustment.
The 0.3 wt% sample of the PHEA-g-PLA self-aggregate solution in D2O were prepared. The small-angle neutron scattering (SANS) measurements were performed using a 9 m SANS HANARO instrument at Korea Atomic Energy Research Institute (KAERI). Neutrons with a wavelength (λ) of 6.38 Å with full width half-maximum Δλ/λ = 11% were used. The sample to detector distance was set to 4.5 m to cover a q-range of 0.008–0.02 Å−1 where q = 4(π/λ)sin(θ/2) is the magnitude of the scattering vector and θ is the scattering angle. The sample scattering was corrected for background and empty cell scattering. The corrected data sets were placed on an absolute scale using standard samples. The SANS data sets were analyzed using a model fit. For the micelles and vesicles, the core–shell model and the hollow sphere model were used, respectively and the core–shell model is described as
 x − xcosx)/x2 where, Δρ is the contrast of the scattering length density (SLD) between the particles and medium and P(q) is an intraparticle interference term (form factor). Furthermore, Vc and Vs are the volumes of the core and shell; Rc and Rs are the radii of the core and shell; and ρc, ρs, and ρsolv are the SLDs of the core, shell, and solvent, respectively, while j1(x) is the first-order spherical Bessel function. In the hollow sphere model, ρc becomes the same as ρsolv. The SLDs of PHEA and PLA used for the SANS data analysis were 1.73 × 10−6 and 1.94 × 10−6 Å−2, respectively. For the SLD calculation, the bulk density of the amorphous PLA (1.248 g cm−3) and molar volume of the repeating hydroxyethyl aspartamide unit (119.5 cm3 mol−1) of PHEA determined using the group contribution method were used.
x − xcosx)/x2 where, Δρ is the contrast of the scattering length density (SLD) between the particles and medium and P(q) is an intraparticle interference term (form factor). Furthermore, Vc and Vs are the volumes of the core and shell; Rc and Rs are the radii of the core and shell; and ρc, ρs, and ρsolv are the SLDs of the core, shell, and solvent, respectively, while j1(x) is the first-order spherical Bessel function. In the hollow sphere model, ρc becomes the same as ρsolv. The SLDs of PHEA and PLA used for the SANS data analysis were 1.73 × 10−6 and 1.94 × 10−6 Å−2, respectively. For the SLD calculation, the bulk density of the amorphous PLA (1.248 g cm−3) and molar volume of the repeating hydroxyethyl aspartamide unit (119.5 cm3 mol−1) of PHEA determined using the group contribution method were used.
The wide-angle X-ray diffraction (WAXD) patterns were obtained uisng a Rigaku D/max-RC (40 kV, 100 mA) using CuKα radiation (λ = 1.542 Å) in a 2θ range from 3° to 40°. The differential scanning calorimetry (DSC) thermograms were obtained at a temperature range from room temperature to 200 °C (Netzsch DSC 204 F1).
2.3. Preparation of larger vesicles
The original precipitation–dialysis method was slightly modified to prepare the larger vesicles. The overall process remained the same except that the solution mixing step was performed in reverse. The DDI water was added dropwise to the PHEA-g-PLA/DMSO solution with vigorous stirring.2.4. Drug loading and release
Calcein as a water-soluble fluorescent probe was entrapped in vesicles using the same original self-aggregate preparation method. The PHEA-g-PLA solution in DMSO (10 mg in 1 mL) was added to 10 mL calcein solution (75 mM) in phosphate-buffered saline (PBS, pH 7.4). The DMSO and non-entrapped calcein were removed during the dialysis against the PBS buffer solution. A burst release of calcein was observed by monitoring the fluorescence emission intensity at 517 nm under the rupture of vesicles by the addition of Triton X-100 (3 wt% as final concentration).To prepare the DOX-loaded vesicles, 4 mg DOX and 10 mg PHEA-g-PLA were dissolved in 1 mL DMSO, which was then added to 10 mL DDI water. The mixture was extensively dialyzed against DDI water. During the dialysis, the water temperature was maintained at 4 °C to prevent the release of the loaded DOX from the vesicles. For the loading assay, the DOX-loaded vesicles were freeze-dried, dissolved in DMSO, and the DOX loading was determined using light absorbance at 482 nm using an ultraviolet/visible (UV/Vis) spectroscopy instrument (Jasco).
2.5. Intracellular delivery of DOX loaded vesicles into cancer cells
HeLa and 293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) at 37 °C in a humidified 5% CO2 atmosphere. The solutions of PHEA-g-PLA and PHEA-g-PLA-(cRGDfC) vesicles loaded with DOX were prepared by dissolving them in PBS solution (pH 7.4) followed by further dilution in serum-free cell-culturing medium. The original cell culturing medium was discarded and replaced with a solution of DOX-loaded PHEA-g-PLA or PHEA-g-PLA–c(RGDfC) vesicles. The DOX loaded in the two different vesicle carriers was adjusted to the same concentration of 1 μg mL−1. After 0.5, 1.5, and 3.0 h incubation, the cells were visualized using a multi-photon confocal laser scanning microscope (CLSM) system (LSM510 META NLO, Carl Zeiss Jena GmbH).The cytotoxicities of the polymer materials, PHEA-g-PLA and PHEA-g-PLA–c(RGDfC) against the HeLa cells were tested using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The polymers were dissolved in PBS solution (pH 7.4), and then diluted in serum-free cell culturing medium to concentrations of 10, 50, 100, and 500 μg mL−1. The HeLa cells were incubated with solutions of the various polymer concentrations for 24 h. The MTT solution was added to each well, incubated for 4 h, the lysis buffer (45% DMF in distilled water and 10% sodium dodecyl sulfate, SDS) was added, and then incubated for 12 h. The light absorbance was measured at 590 nm using a microplate reader (Bio-Rad, Model 550) and the fraction of surviving cells was determined. The viability of the HeLa cells incubated with DOX-loaded PHEA-g-PLA vesicles, DOX-loaded PHEA-g-PLA-(cRGDfC) vesicles, and free DOX was determined using the same protocol. The concentration of the free DOX and DOX loaded in both vesicles was adjusted to a consistent 5 or 10 μM.
3. Results and discussion
A series of five PHEA-g-PLA comb-like graft copolymers with a wide range of different hydrophilic/hydrophobic ratios was synthesized. PLA is a hydrolytic degradable aliphatic polyester while PHEA is a water-soluble synthetic poly(amino acid) and, therefore, is an excellent in vivo-degradable and – absorbable alternative to PEGs.22 Table 1 summarizes the molecular characterization of the PHEA-g-PLA copolymers. The PHEA modified to contain the minimal number of primary amine groups (PHEA-NH2) was prepared as the water-soluble backbone polymer. The PHEA-NH2 (degree of polymerization, DP = 531 (ref. 23)) was verified using 1H-NMR and consisted of hydroxyethyl aspartamide units (88%) as the major backbone polymer and aminoethyl aspartamide units (12%) for the grafting reaction with PLA. The reactions for synthesizing PLA and modifying the distal hydroxyl group of PLA with CDI (PLA-CDI) were successful. The MW and DP of the PLA were calculated as 2456 and 32, respectively based on the 1H-NMR spectra. The MW determined as 2412 using MALDI-TOF mass spectroscopy was similar to the MW calculated from the 1H-NMR spectra. At the grafting step, the feeding amount of PLA-CDI for the repeating PHEA-NH2 units was varied from 2.5 to 12.5 mol%. The degree of substitution (DS), which represented the grafting density as grafted mole% was calculated based on the 1H-NMR spectra and elemental analysis. The grafting reaction was successful with a reaction efficiency > 60% that yielded a DS of 1.4–9.2. The weight and volume fractions of the hydrophobic PLA portion and the hydrophilic backbone polymer were easily calculated from the DS values. The volume fraction of the hydrophilic backbone, fHPL(v) was controlled from 0.39 to 0.81. In addition to the straightforward molecular composition control, the PHEA-g-PLA copolymer platform can be favorably upgraded to a multivalent or multifunctional system using abundant hydroxyl and residual amino groups.| PHEA–PLA1.4 | PHEA–PLA3.0 | PHEA–PLA4.4 | PHEA–PLA6.3 | PHEA–PLA9.2 | |
|---|---|---|---|---|---|
| a Degree of substitution of PLA determined based on 1H-NMR spectra. DS values calculated from elemental analysis results are similar as 1.43, 3.17, 4.30, 6.59, and 9.11.19b Number of PLA segments per one PHEA-g-PLA copolymer molecule. No. of PLA = DS/100 × DP of backbone polymer (531).c Volume fraction of hydrophilic backbone polymer of PHEA-g-PLA copolymer molecule. fHPL(v) values were calculated based on DS, bulk density of amorphous PLA (1.248),23 and molar volume of hydroxyethyl aspartamide unit (119.5 cm3 mol−1 calculated with group contribution theory24). | |||||
| Feed mole% | 2.5 | 5.0 | 7.5 | 10.0 | 12.5 |
| DSa | 1.38 | 2.98 | 4.37 | 6.34 | 9.16 |
| No. of PLAb | 7 | 16 | 23 | 34 | 49 |
| fHPL(v)c | 0.81 | 0.67 | 0.58 | 0.48 | 0.39 |
The size, structure, and thermodynamic stability of the self-aggregates of the PHEA-g-PLA copolymers were thoroughly examined (Table 2). The size and structure of the self-aggregates of all five PHEA-g-PLA copolymers were first characterized using DLS and TEM.19 PHEA–PLA1.4 and PHEA–PLA3.0 formed mono-disperse spherical micelles with a diameter approximately 20 nm (Fig. 1 and S1†). PHEA–PLA6.3 and PHEA–PLA9.2 formed monodisperse unilamellar vesicles with a of diameter approximately 40 nm (Fig. 1b). The characterization of PHEA–PLA4.4 identified two separate size distribution populations using DLS and two distinct structures, micelles and vesicles were observed in the TEM images. The structure of the self-aggregates was controlled as a function of the grafting degree of PLA (DS), which affects both chemical composition and the geometrical aspects of copolymer molecules. The structural transition of the PHEA-g-PLA self-aggregates can be described by the molecular packing parameter (p) concept. It is well known that p is defined as v0/al0, where v0 and l0 are the volume and the length of the hydrophobic group of amphiphilic molecule, respectively, and a is the surface area of the hydrophobic core of the self-aggregate expressed per amphiphilic molecule.24 Presently, the complex geometry of a graft copolymer molecule can be simplified as the composition of the connected multiple basic building units.25 By applying the concept of dividing a graft copolymer molecule into the multiple pieces based on the number of grafted chains, a basic building unit can be defined as a piece of the copolymer consisting of a segment of PHEA backbone grafted with one PLA chain. Since the hydrophobic PLA chain remained constant in all five PHEA-g-PLA copolymers, the ratio “v0/l0” is also constant regardless of the DS, whereas “a” is decreased when the increased DS divides the copolymer molecule into multiple pieces with a shorter backbone segment. Overall, the increase in DS increases the packing parameter and, thus, leads to the structural transition from micelles to vesicles (Table 3).
| PHEA–PLA1.4a | PHEA–PLA3.0 | PHEA–PLA4.4 | PHEA–PLA6.3 | PHEA–PLA9.2a | |
|---|---|---|---|---|---|
| a Samples proceeded to SANS experiments.b Structure of self-aggregates confirmed with TEM images.c Critical aggregation concentration determined using pyrene as a fluorescent probe.d Average values from three DLS measurements.e Larger vesicles with average diameter of 145.1 ± 4.7 were able to be prepared by modified precipitation–dialysis method.f Larger vesicles with average diameter of 156.9 ± 4.7 nm or giant vesicles with size of 4–5 μm were able to be prepared by modified precipitation–dialysis or film-hydration method, respectively.19g Nagg_LA: aggregation number of PLA segments per one self-aggregate particle. Nagg: aggregation number of PHEA-g-PLA copolymer chains per one self-aggregate particle. | |||||
| Structureb | Spherical micelles | Spherical micelles | Coexistence of micelles and vesicles | Vesicles | Vesicles |
| CACc (μg mL−1) | 22.4 | 4.9 | 4.6 | 2.9 | 1.9 |
| Diameterd (nm) | 22.3 ± 1.2 | 16.2 ± 1.1 | 36.1 ± 3.0 | 41.2 ± 3.3e | 43.2 ± 2.8f |
| Polydispersityd | 0.073 ± 0.017 | 0.091 ± 0.008 | 0.115 ± 0.009 | 0.083 ± 0.005 | 0.080 ± 0.013 |
 | ||
| Fig. 1 TEM images of negatively stained (a) PHEA–PLA1.4 micelles and (b) PHEA–PLA9.2 vesicles. Scale bars represent 40 nm. | ||
| USP(DOX) | cRGD-USP(DOX) | |
|---|---|---|
| a Determined based on 1H-NMR spectra.b Number of c(RGDfC) peptides per one PHEA-g-PLA–c(RGDfC) copolymer molecule. No. of c(RGDfC) = DS/100 × DP of backbone polymer (531).c Determined by UV/Vis absorbance.d Average values from three DLS measurements. | ||
| PHEA-g-PLA copolymer | PHEA–PLA6.3 | PHEA–PLA6.3 conjugated with c(RGDfC) |
| DS of c(RGDfC)a | — | 0.56 |
| No. of c(RGDfC)b | — | 3 |
| DOX contentc (wt%) | 4.2 | 4.4 |
| Loading efficienty (%) | 14.0 | 14.7 |
| Diameterd (nm) | 44.1 ± 5.7 | 45.3 ± 6.0 |
| Polydispersityd | 0.085 ± 0.011 | 0.079 ± 0.015 |
Although the above discussion explains the overall trend of the self-aggregate structures adequately, the application of the molecular packing parameter has limitations. The assumption of graft copolymer molecule as connected multiple basic building units may be valid only from a geometrical point of view, for the thermodynamic functions of present nonlinear graft copolymer system. In particular, the configurational entropies are different from those of the surfactants or linear block copolymers owing to the covalent segmental connection. The segmental connection would influence the flexibility of the backbone chain, which was recognized as an important additional factor for the self-assembly of graft copolymers after being first reported in our previous studies.20,26–28 The more PLA chains are grafted, the stiffer the backbone chain with the crowded bulky side chains becomes. In addition, it becomes less prone to bending to localize the grafted PLA chains in a small spherical dimension. The copolymer molecules would rather form a bilayer structure with a low backbone curvature than a micellar core enclosed with highly curved backbones. The present PHEA-g-PLA system started to form vesicles when the volume fraction of the hydrophilic part was <0.58, which is a marked deviation from the value generally associated with PEG-based block copolymer systems (fPEG(v) < 0.4).29,30 This deviation is likely attributable to the flexibility and bending behavior of the backbone chain as a unique topological factor of the nonlinear graft polymers, compared to the inflexible linear block copolymers.
To analyze the internal construction of the two distinct nanostructures (micelles and vesicles), the self-aggregates of PHEA–PLA1.4 and PHEA–PLA9.2 were selected for further investigation using SANS, WAXD, and DSC. The nanostructural dimensions of the PHEA–PLA1.4 micelles and PHEA–PLA9.2 vesicles were quantifiable using the SANS studies.3,31,32 The SANS experimental data were analyzed using curve fitting with a core–shell sphere model for the micelles and a hollow sphere model for the vesicles. Fig. 2a shows the SANS results of the PHEA–PLA1.4 self-aggregates modeled as core–shell spheres to describe their micellar structure, which consisted of a core region composed of PLA and a PHEA shell. The model fit agreed with the SANS intensity very well, resulting in a core radius (Rc) of 8.04 nm and a shell thickness (δ) of 3.57 nm. Fig. 2b shows the SANS experimental data of the PHEA–PLA9.2 vesicles and the model-fitted curve. The experimental points were in perfect agreement in the q-range with the fitted curve using the hollow sphere form factor, which represented the fluid-filled internal compartment as a hollow core and the enclosing PLA bilayer as a shell. The core radius, Rc and shell thickness, δ were determined as 13.63 and 9.08 nm, respectively. Additionally, the q−2 behavior in the low q region indicated the existence of a bilayer membrane. The SANS analysis corroborated the DLS and TEM results, which led to the general acceptance of the smaller dimensions that appeared in the TEM observation in vacuo. The light and neutron scattering measurements and electron microscopy images all supported the formation of well-organized micellar and vesicular nanostructures by the PHEA-g-PLA copolymers in relation to the DS of PLA. The aggregation number of PLA (Nagg_PLA), which is the number of PLA chains forming one self-aggregate can be calculated from the structural information from the SANS analysis using the following equations where the volume of one hydrophobic PLA domain is divided by the molecular volume of one PLA chain.
| Nagg_PLA(PHEA–PLA1.4) = 4πρPLARc3NA/3MnPLA |
| Nagg_PLA(PHEA–PLA9.2) = 4πρPLA((Rc + δ)3 − Rc3)NA/3MnPLA |
| Nagg(PHEA–PLA1.4) = 4πρPLARc3NA/3MnPLA/(DPbackbone × DSPHEA–PLA1.4/100) |
| Nagg(PHEA–PLA9.2) = 4πρPLA((Rc + δ)3 − Rc3)NA/3MnPLA/(DPbackbone × DSPHEA–PLA9.2/100) |
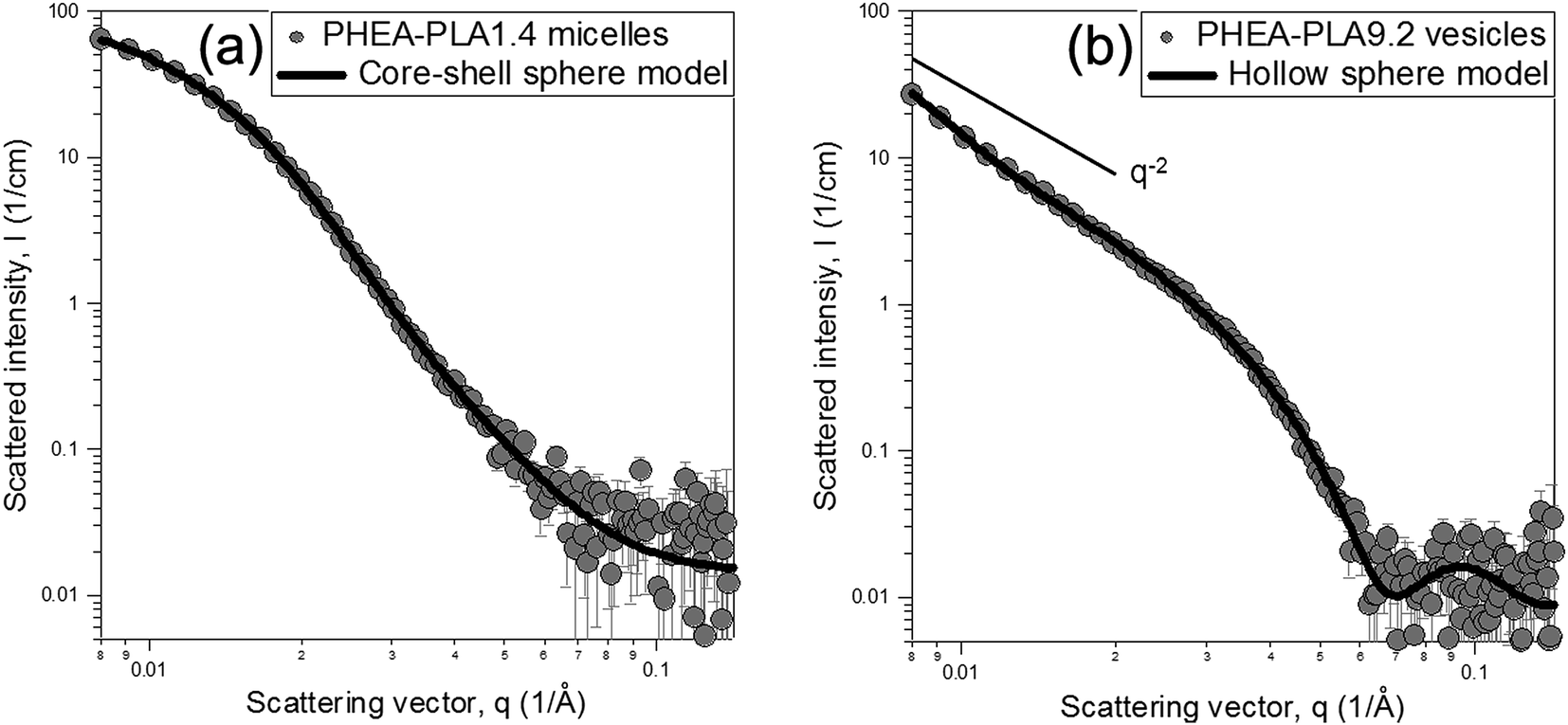 | ||
| Fig. 2 SANS experimental data sets and the model-fitted curves of (a) PHEA–PLA1.4 micelles and (b) PHEA–PLA9.2 vesicle in D2O. | ||
For the PHEA–PLA1.4 micelles, the Nagg_PLA and Nagg were calculated as 680 and 93, respectively while those for PHEA–PLA9.2 vesicles were 12004 and 247, respectively.
The thickness and molecular structure of the polymersome bilayer are important factors affecting the properties of vesicles including their stability. The bilayer structure of the PHEA–PLA9.2 vesicles can be described in relation to the bilayer thickness determined using the SANS experiments. The MW and chain conformational state of the bilayer-forming hydrophobic segments are decisive factors affecting the bilayer thickness (d). The empirical relation is known as d ∼ (MWHPB)a where, MWHPB is the MW of the bilayer-forming hydrophobic segment and a is 1 or 0.5 for the fully stretched or complete random coil conformation, respectively.33,34 Since the PHEA-g-PLA system was designed without varying the PLA MW and no comparable graft copolymers was reported, the PEO-b-PLA system was used for the comparison. This is acceptable because the bilayer thickness or structure is dominated by hydrophobic segments. Disher et al. reported that PEO-b-PLA vesicles with MW of PLA as 3200 or 4000 formed 10.4 or 11.4 nm-thick bilayers, respectively.11,30 Scaling the MW of PLA and the d of those examples showed that the a was 0.5, which implies that the PLA chains of PEO-b-PLA copolymers showed a random coil conformation within the bilayer domain. The 9.08 nm thick bilayer of the PHEA–PLA9.2 vesicles with PLA at MW 2412 exactly matched the trend of a = 0.5 of PEO-b-PLA system and, therefore, the represented PLA segments of PHEA–PLA9.2 were randomly coiled in the bilayer domain. As expected, the bilayer region dominated by the hydrophobic segment with no significant involvement of hydrophilic chain showed the same molecular conformation as the hydrophobic segment regardless of the copolymer architectures.
The molecular conformation of PLA as well as the overall ordering state of the hydrophobic PLA domains of the self-aggregates were investigated using WAXD and DSC analyses. Fig. 3a shows the WAXD profiles of the homopolymer PLA (PHEA–PLA1.4 bulk polymer) that did not go through the self-assembly process as well as those of the freeze-dried PHEA–PLA1.4 micelles and PHEA–PLA9.2 vesicles. The homopolymer PLA showed a crystalline structure with the clear characteristic peaks of PLA. The bulk copolymer, PHEA–PLA1.4 showed all the same peaks that appeared in the profile of the homopolymer PLA. The long-range high ordering of PLA segments was well maintained in the PHEA-g-PLA before the self-assembly occurred. The profiles of both freeze-dried PHEA–PLA1.4 micelles and PHEA–PLA9.2 vesicles showed that they almost completely lost their crystallinity. During self-assembly, the PLA segments of the PHEA-g-PLA copolymers were reoriented randomly into an amorphous state in the intra-aggregate PLA domains. The DSC thermograms in Fig. 3b are consistent with the WAXD results. The homopolymer PLA and PHEA–PLA1.4 bulk copolymer exhibited the thermal behavior of crystal polymers and showed only a melting temperature (Tm). In contrast, the PHEA–PLA1.4 micelles and PHEA–PLA9.2 vesicles exhibited the thermal behavior of amorphous polymers and showed a glass transition temperature (Tg), recrystallization, and Tm. The amorphous hydrophobic domains of the self-aggregates of the PHEA-g-PLA, micelle core and vesicle bilayer with random coiled PLA segments, are expected to provide favorable environment into which the hydrophobic drug molecules can be well mixed.
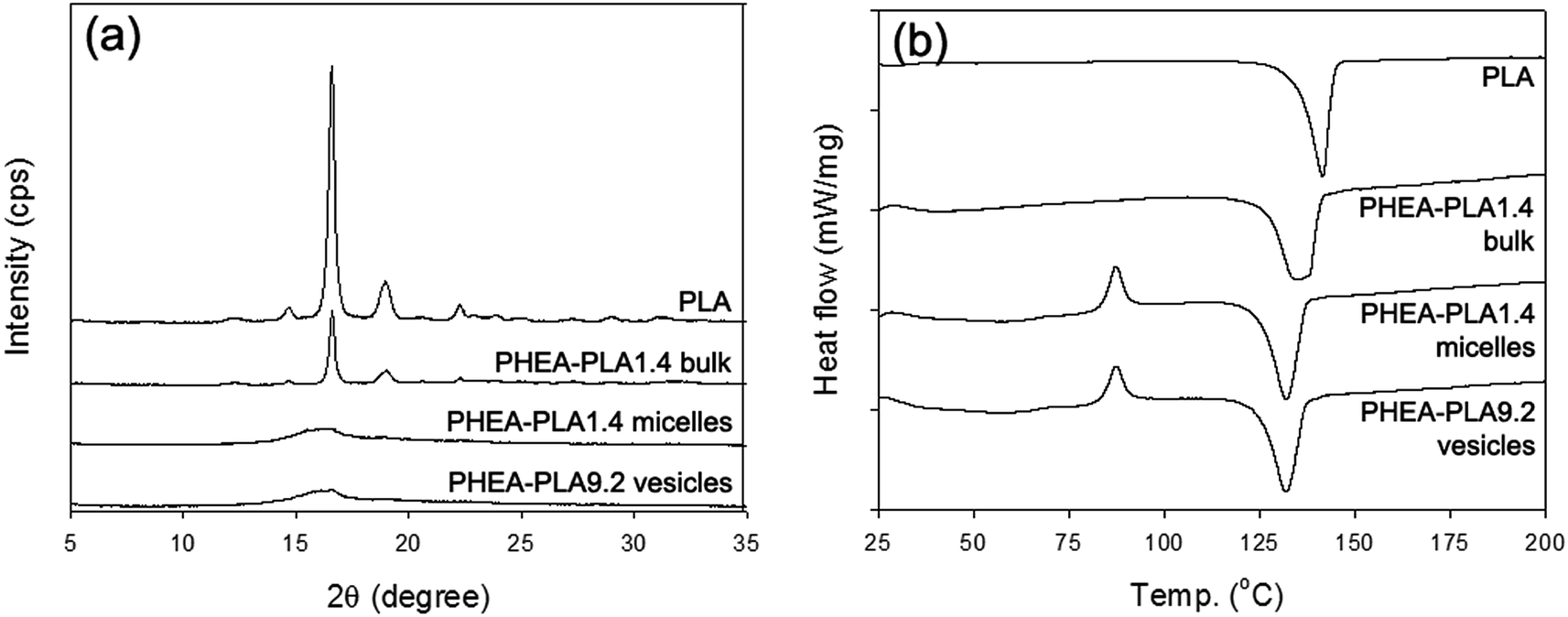 | ||
| Fig. 3 (a) WAXD profiles and (b) DSC thermograms of PLA homopolymer, PHEA–PLA1.4 copolymer never gone through self-assembly process, freeze-dried PHEA–PLA1.4 micelles, and freeze-dried PHEA–PLA9.2 vesicles. | ||
The CAC, which represents the thermodynamic stability of the self-aggregates was measured using pyrene as shown in Fig. 4a. The CACs of PHEA-g-PLA copolymers were determined as 22.4–1.9 μg mL−1, which are as low at only several to several tens of nanomolar (Fig. 4b). In addition to the high thermodynamic stability, the strong intra-/inter-associational characteristics of the graft copolymers is presumed to contribute to their kinetic stability. Because the copolymer has a high DS of PLA, the CAC was reduced. However, the previously known linear relationship between the CAC and DS (logCAC = A − B × DS) discovered for graft copolymer micelles was not obtained in this study.25,35 The rapidly decreasing trend observed in the micellar aggregate region between PHEA–PLA1.4 and PHEA–PLA3.0, was not maintained in the subsequent PHEA–PLA4.4 polymer, which had a composition that started to form vesicles. This nonlinear trend of CAC decrease and change of slope at the structure transitional region can be an indicator of the structural transition of the graft copolymer self-aggregates.
 | ||
| Fig. 4 (a) Determination of CAC of PHEA–PLA9.2 with the plot of intensity ratio (I336/I333) of pyrene excitation spectra vs. polymer concentration. (b) Change of CACs according to DS of PLA. | ||
Numerous recent studies have demonstrated that the whole body and cellular distribution of nanoscale drug carriers is influenced by the size and morphology of the drug carriers. Therefore, the ability to control or adjust these properties in drug carriers is advantageous for optimizing drug delivery systems. In the case of the polymeric self-aggregates, their structure is mainly determined by the hydrophilic/hydrophobic ratio of the polymer molecules. The self-aggregate preparation method as a self-assembly pathway is a very important factor in determining the particle size. Especially, the size of polymersomes can vary considerably from several tens of nanometers to several tens of micrometers.36–38 It has been shown that ultrasmall vesicle-forming graft copolymers can also self-assemble into giant vesicles with a size of 4–5 μm using the film hydration method.20 Here, to alter the vesicle size to within a narrow nanoscopic range, we decided to modify the original precipitation–dialysis method, which yielded ultrasmall vesicles rather than using an entirely different preparation method. Since vesicles are formed during the solution-mixing step when the PHEA-g-PLA copolymers are exposed to the aqueous medium, the thermodynamic condition of the solution mixing process is a size-determining factor. Modifying the solution mixing process could change the enthalpy/entropy balance, which controls the growth of self-aggregates, to generate a finite size and aggregation number. The mixing process of the DDI water and PHEA-g-PLA copolymer solution in DMSO was simply reversed. The addition of DDI water to the polymer solution in DMSO induced the formation of vesicles with a diameter of approximately 150 nm. Fig. 5a shows the TEM images of vesicles that were >100 nm with a clear unilamellar vesicular structure, and showed no change in the bilayer thickness compared to the ultrasmall vesicles. The monodisperse size distribution in the range of 100–200 nm revealed by the DLS correlated closely with the TEM results (Fig. 5b). In the good-solvent-rich and copolymer-concentrated environment of the modified method, the copolymer molecules exhibited a relatively low interfacial energy and existed in a crowded state. The resultant larger vesicles with a presumably larger aggregation number imply a higher enthalpic gain and entropic loss, compared to that of the ultrasmall vesicles from the selective-solvent-rich and copolymer-dilute environment of the original method. These effects are consistent with what was observed in other studies including the simulation of surfactant molecules assembled into micelles with a different size and aggregation number, and the solvent effects on the self-assembly of hydrophobic solutes.39,40 Therefore, it is obvious that the ultrasmall size of the vesicles was induced by this particular self-assembly process using the precipitation–dialysis method. In addition, the process was influenced by the molecular nature of the PHEA-g-PLA graft copolymer. The ability to form different-sized vesicles by minor changes in the preparation method facilitates the exploration of the size effect and optimization of the drug delivery system.
 | ||
| Fig. 5 (a) TEM images of negatively stained larger vesicles of PHEA–PLA9.2. Both scale bars represent 100 nm. (b) Size distribution of larger vesicles of PHEA–PLA9.2 in comparison with that of ultrasmall vesicles of PHEA–PLA9.2.19 | ||
In addition, we designed cancer-targeted drug delivery systems based on the ultrasmall vesicles. DOX was selected as a water-soluble anticancer therapeutic agent to be loaded into the inner hydrophilic compartment of the vesicles. First, the ability of the vesicles to entrap the water-soluble molecules into the aqueous media-filled internal space was confirmed using calcein, which is a hydrophilic fluorescent probe as the model molecule. The calcein-entrapped vesicles were easily prepared using the same original precipitation–dialysis method, and the only difference was that the DDI water was substituted with the calcein solution. The effective loading ability was demonstrated by the large difference in the fluorescence emission intensity of calcein between the entrapped and totally released state (Fig. 6a). In the entrapped state, the calcein was tightly localized in a restricted space and its fluorescence intensity was as low as that of the background level due to the fluorescence quenching effect. After the vesicles rupture following exposure to Triton X-100, the calcein bursting out of disassembling vesicles and showed a rapidly increased fluorescence intensity. The calcein loading experiment showed that the PHEA-g-PLA ultrasmall vesicles can be used as carriers for water-soluble bioactive substances. The PHEA–PLA6.3 and PHEA–PLA9.2 vesicles with similar sizes also showed comparable calcein loading capacities.
 | ||
| Fig. 6 (a) Bursting release of entrapped calcein under dissociation of vesicles. (b) Release patterns of DOX from PHEA–PLA6.3 vesicles at 37 °C. | ||
The PHEA–PLA6.3 vesicles used in the further experiments were loaded with DOX using the same precipitation–dialysis method, except that the DDI water was substituted with the DOX-dissolved aqueous solution. The entrapment and release of DOX were spectrophotometrically quantified. DOX-loaded PHEA–PLA6.3 ultrasmall vesicles with a size <50 nm without any significant size change were successfully prepared. A 2.0–4.5 wt% DOX content was obtained based on a DOX feeding amount of 20–40 wt% to the copolymer amount. The in vitro release of DOX from the PHEA–PLA6.3 vesicle carriers loaded with 4.2 wt% DOX was monitored. Two different pH conditions (pH 7.4 and pH 5.0) were used as the physiological and early endosomal pH, which drug carriers would be exposed to following intravenous administration and cellular uptake, respectively. Fig. 6b shows the pH-dependent release pattern of DOX. The DOX molecules were released faster at pH 5.0 than they were at pH 7.4. The pH-dependent release pattern is attributable to the pH-sensitivity of the DOX molecule, which contains an amino group and the faster degradation of the ester bonds of PLA and carbamate linkage between the backbone and PLA at acidic pH. The PHEA-g-PLA vesicles released the drug faster under the early endosomal pH condition than they did under the physiological pH condition, which would facilitate the sustained drug release process during circulation in blood stream and accelerated drug release after cellular internalization.
For effective intracellular delivery of therapeutics to cancer cells, cyclic RGD peptide was introduced as an active targeting moiety that binds to αvβ3 and αvβ5 integrins overexpressed on various cancer or angiogenic blood vessel cells. The cyclic RGD peptide with a sequence of c(RGDfC) was conjugated to the PHEA–PLA6.3 copolymer through a heterobifunctional coupling reagent containing short PEG spacer, NHS-EO2-maleimide, which links the residual amines of the PHEA-g-PLA copolymer backbone and the thiol of cysteine in the c(RGDfC) as shown in Scheme 1. The 1H-NMR spectra in DMSO-d6 reveal the successful modification of the PHEA-g-PLA with the c(RGDfC) peptide (Fig. 7b). As summarized in Table S2,† the PHEA-g-PLA–c(RGDfC) copolymer molecules were characterized to have three c(RGDfC) peptides on one copolymer chain based on the DS of c(RGDfC) calculated as 0.56. The 1H-NMR spectra in D2O proved the formation of self-aggregates with a solid-like hydrophobic PLA domain and outward exposure of the cyclic RGD peptides to aqueous medium. The TEM analysis confirmed the structure of the PHEA-g-PLA–c(RGDfC) self-aggregates as ultrasmall unilamellar vesicles that were similar to the PHEA-g-PLA copolymer vesicles (Fig. 7c). The DOX-entrapped ultrasmall vesicle carriers formulated from the PHEA–PLA6.3 and cyclic RGD peptide-conjugated PHEA–PLA6.3, were named USP(DOX) and cRGD-USP(DOX), respectively, and they show similar sizes and DOX contents. Therefore, it noteworthy that the conjugation of the cyclic RGD peptide with low DS did not significantly change the physicochemical properties or hamper the formation of a well-defined vesicular structure.
 | ||
| Scheme 1 Synthetic route for PHEA-g-PLA-g-c(RGDfC) from PHEA-g-PLA. | ||
 | ||
| Fig. 7 (a) Molecular formula of PHEA-g-PLA–c(RGDfC). (b) 1H-NMR spectra of PHEA-g-PLA and PHEA-g-PLA–c(RGDfC) in DMSO-d6 (top) and in D2O (bottom). (c) TEM image of negatively stained PHEA-g-PLA–c(RGDfC). Scale bar represents 40 nm. | ||
The USP(DOX) and cRGD-USP(DOX) were incubated with HeLa cells overexpressing αvβ3 and αvβ5 integrins, which can be recognized by the cyclic RGD peptide moiety. Fig. S2† shows the CLSM images of HeLa cells incubated with USP(DOX) and cRGD-USP(DOX), respectively. The cellular uptake of the vesicle carriers was visualized based on the fluorescence emitted by the DOX. The CLSM study showed that the cRGD-USP(DOX) was more highly internalized in the HeLa cells than the USP(DOX) was at every incubation time point. The fluorescent intensity of the cells incubated with cRGD-USP(DOX) was stronger than that of the USP(DOX)-incubated cells was. In addition, cells incubated with cRGD-USP(DOX) showed a more obvious fluorescence from the cell membranes than the USP(DOX)-incubated cells did. The cRGD-USP(DOX) appeared to effectively bind to the HeLa cells surface and enhanced the intracellular drug delivery. We also noted that the control USP(DOX) showed obvious cellular uptake, although it was not as much as that of the cRGD-USP(DOX) was. Therefore, it is expected that the USP(DOX) would be adequately internalized into cancer cells once it penetrates the target cancer tissue by circumventing the reticuloendothelial system (RES) and renal clearance. In addition, the USP(DOX) would likely be internalized into the cells more effectively following its functionalized with a targeting moiety such as the cyclic RGD peptide. The active targeting of the vesicles to the HeLa cells based on selective recognition between cyclic RGD peptide and αvβ3 and αvβ5 integrins was corroborated by experiments with 233T cells as the negative control cell line that expressed only αvβ5 integrins, which showed a 10-fold lower binding affinity for cyclic RGD peptide than that of the αvβ3 integrin.41,42 No significant difference in strength and pattern of fluorescence was observed between the 293T cells incubated with USP(DOX) or cRGD-USP(DOX).
The noncytotoxicity of the polymer materials, PHEA-g-PLA and PHEA-g-PLA–c(RGDfC), was confirmed using an MTT assay as shown in Fig. S3a.† Then, the anticancer chemotherapeutic effects of USP(DOX) and cRGD-USP(DOX) against the HeLa cells were measured and compared with that of the free DOX as shown in Fig. S3b.† Both USP(DOX) and cRGG-USP(DOX) demonstrated recognizable cytotoxicity, although their effects were not as potent as that of the free DOX. The cancer cell cytotoxicity results of the USP(DOX) and cRGD-USP(DOX) were consistent with the cellular uptake results discussed earlier. The cRGD-USP(DOX) showed a higher cellular internalization than USP(DOX) did and, therefore, a higher anticancer therapeutic effect was observed in the cRGD-USP(DOX)-treated cells. The active cancer-targeting effect can be more improved by increasing the DS of the cyclic RGD peptide or increasing the length of the space between the backbone and cyclic RGD peptide. In summary, the cell experiments suggest that PHEA-g-PLA is biocompatible and its ultrasmall polymersomes can deliver DOX and induce anticancer cell cytotoxicity. In addition, the intracellular delivery and anticancer effect of the vesicles were enhanced by the functionalization of the polymersomes with an active targeting moiety.
4. Conclusion
In summary, a series of amphiphilic biodegradable graft copolymers (PHEA-g-PLA) with a wide range of hydrophilic/hydrophobic ratios obtained by varying the grafting degree of PLA was synthesized. The PHEA-g-PLA copolymers self-assembled into micelles with a 20 nm diameter or vesicles with a 40 nm as a function of the PLA grafting degree. The self-aggregate structural transition was based on the combination of a classical packing parameter concept and the flexibility of the backbone as an additional topological factor of the graft copolymer molecule. The CAC values not only suggested a high thermodynamic stability, but they also indicated the structural transition of the self-aggregates. Both the micelles and vesicles showed well-defined uniform nanostructures, consisting of a compartment for drug loading and a protective layer. The vesicles were targeted as drug carriers for water-soluble therapeutic agents and potential multiple drug delivery systems. The vesicles were able to entrap the water-soluble DOX and released it in an acid-sensitive manner. The ease of functionalization was demonstrated by the successful introduction of the cyclic RGD peptide for active cancer-targeting. Furthermore, the vesicle size was altered to a nanoscopic range by a simple modification of the self-assembly route, which manipulated the thermodynamic balance. The present ultrasmall polymersomes can be considered as a potential general drug delivery platform, which could also be used to explore the size effect of polymersome drug carriers. In addition, these polymersomes could be used to investigate the structural effect of various nanoscale drug carriers with similar sizes including polymer micelles, polymer nanoparticles, dendrimer clusters, and even inorganic nanoparticles.Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) Grant funded by the Korean Government, Ministry of Science, ICT and Future Planning (MSIP, No. 2016M2B2A4910207).Notes and references
- H. Qiu, Z. M. Hudson, M. A. Winnik and I. Manners, Science, 2015, 347, 1329 CrossRef CAS PubMed.
- H. Qiu, Y. Gao, V. A. Du, R. Harniman, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2015, 137, 2375 CrossRef CAS PubMed.
- N. P. Truong, J. F. Quinn, M. V. Dussert, N. B. T. Sousa, M. R. Whittaker and T. P. Davis, ACS Macro Lett., 2015, 4, 381 CrossRef CAS.
- V. Kapishon, R. A. Whitney, P. Champagne, M. F. Cunningham and R. J. Neufeld, Biomacromolecules, 2015, 16, 2040 CrossRef CAS PubMed.
- G. K. Such, Y. Yan, A. P. R. Johnston, S. T. Gunawan and F. Caruso, Adv. Mater., 2015, 27, 2278 CrossRef CAS PubMed.
- W. Park, B.-C. Bae and K. Na, Biomaterials, 2016, 77, 227 CrossRef CAS PubMed.
- M. Kanamala, W. R. Wilson, M. Yang, B. D. Palmer and Z. Wu, Biomaterials, 2016, 85, 152 CrossRef CAS PubMed.
- N. J. Warren, O. O. Mykhaylyk, A. J. Ryan, M. Williams, T. Doussineau, P. Dugourd, R. Antoine, G. Portale and S. P. Armes, J. Am. Chem. Soc., 2015, 137, 1929 CrossRef CAS PubMed.
- W. Jiang, Y. Zhou and D. Yana, Chem. Soc. Rev., 2015, 44, 3874 RSC.
- M. Hu, Y. Shen, L. Zhang and L. Qiu, Biomacromolecules, 2016, 17, 1026 CrossRef CAS PubMed.
- F. Ahmed, R. I. Pakunlu, G. Srinivas, A. Brannan, F. Bates, M. L. Klein, T. Minko and D. E. Discher, Mol. Pharmaceutics, 2006, 3, 340 CrossRef CAS PubMed.
- S. F. M. van Dongen, W. P. R. Verdurmen, R. J. R. W. Peters, R. J. M. Nolte, R. Brock and J. C. M. van Hest, Angew. Chem., Int. Ed., 2010, 49, 7213 CrossRef PubMed.
- X. Yang, J. J. Grailer, I. J. Rowland, A. Javadi, S. A. Hurley, V. Z. Matson, D. A. Steeber and S. Gong, ACS Nano, 2010, 4, 6805 CrossRef CAS PubMed.
- J. S. Lee, W. Zhou, F. Meng, D. Zhang, C. Otto and J. Feijen, J. Controlled Release, 2010, 146, 400 CrossRef CAS PubMed.
- A. Lavasanifar, J. Samuel and G. S. Kwon, Adv. Drug Delivery Rev., 2002, 54, 169 CrossRef CAS PubMed.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249 CrossRef CAS PubMed.
- S. E. A. Gratton, P. A. Ropp, P. D. Pohlhaus, J. C. Luft, M. E. N. Madden and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11613 CrossRef CAS PubMed.
- F. Osaki, T. Kanamori, S. Sando, T. Sera and Y. Aoyama, J. Am. Chem. Soc., 2004, 126, 6520 CrossRef CAS PubMed.
- W. Jiagn, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Nat. Nanotechnol., 2008, 3, 145 CrossRef PubMed.
- H. J. Lee, S. R. Yang, E. J. An and J.-D. Kim, Macromolecules, 2006, 39, 4938 CrossRef CAS.
- J. Thiele, D. Steinhauser, T. Pfohl and S. Förster, Langmuir, 2010, 26, 6860 CrossRef CAS PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288 CrossRef CAS PubMed.
- J. H. Jeong, H. S. Kang, S. R. Yang and J.-D. Kim, Polymer, 2003, 44, 583 CrossRef CAS.
- J. N. Israelachvili, Intermolecular and Surface Forces, Academic Press, San Diego, CA, 2nd edn, 1992 Search PubMed.
- S. Förster, M. Zisenis, E. Wenz and M. Antonietti, J. Chem. Phys., 1996, 104, 9956 CrossRef.
- L. Zhang, J. Lin and S. Lin, J. Phys. Chem. B, 2007, 111, 9209 CrossRef CAS PubMed.
- H. Qi and C. Zhong, J. Phys. Chem. B, 2008, 112, 10841 CrossRef CAS PubMed.
- C. Cai, J. Lin, T. Chen and X. Tian, Langmuir, 2010, 26, 2791 CrossRef CAS PubMed.
- Y.-Y. Won, A. K. Brannan, H. T. Davis and F. S. Bates, J. Phys. Chem. B, 2002, 106, 3354 CrossRef CAS.
- F. Ahmed and D. E. Discher, J. Controlled Release, 2004, 96, 37 CrossRef CAS PubMed.
- H. Kukula, H. Schlaad, M. Antonietti and S. Förster, J. Am. Chem. Soc., 2002, 124, 1658 CrossRef CAS PubMed.
- F. Chécot, A. Brúlet, J. Oberdisse, Y. Gnanou, O. Mondain-Monval and S. Lecommandoux, Langmuir, 2005, 21, 4308 CrossRef.
- H. Bermudez, A. K. Brannan, D. A. Hammer, F. S. Bates and D. E. Discher, Macromolecules, 2002, 35, 8203 CrossRef CAS.
- G. Battaglia and A. J. Ryan, J. Am. Chem. Soc., 2005, 127, 8757 CrossRef CAS PubMed.
- H. S. Kang, S. R. Yang, J.-D. Kim, S.-H. Han and I.-S. Chang, Langmuir, 2001, 17, 7501 CrossRef CAS.
- M. Antonietti and S. Förster, Adv. Mater., 2003, 15, 1323 CrossRef CAS.
- C. LoPresti, H. Lomas, M. Massignani, T. Smart and G. Battaglia, J. Mater. Chem., 2009, 19, 3576 RSC.
- J. Dua and R. K. O'Reilly, Soft Matter, 2009, 5, 3544 RSC.
- C. M. Care and T. Dalby, Europhys. Lett., 1999, 45, 38 CrossRef CAS.
- N. Choudhury and B. M. Pettitt, J. Phys. Chem. B, 2006, 110, 8459 CrossRef CAS PubMed.
- M. Oba, K. Aoyagi, K. Miyata, Y. Matsumoto, K. Itaka, N. Nishiyama, Y. Yamasaki, H. Koyama and K. Kataoka, Mol. Pharmaceutics, 2008, 5, 1080 CrossRef CAS PubMed.
- M. Oba, S. Fukushima, N. Kanayama, K. Aoyagi, N. Nishiyama, H. Koyama and K. Kataoka, Bioconjugate Chem., 2007, 18, 1415 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra13675c |
| This journal is © The Royal Society of Chemistry 2016 |