
Manganese silicate drapes as a novel electrode material for supercapacitors†
Hong-Yan Wang,
Yue-Ya Wang,
Xue Bai,
Huan Yang,
Jian-Ping Han,
Ning Lun,
Yong-Xin Qi* and
Yu-Jun Bai*
Key Laboratory for Liquid–Solid Structural Evolution and Processing of Materials (Ministry of Education), Shandong University, Jinan 250061, PR China. E-mail: byj97@126.com; qyx66@sdu.edu.cn
First published on 1st November 2016
Abstract
Manganese silicate was fabricated via a facile hydrothermal reaction between Na2SiO3·9H2O and MnCl2·4H2O. The morphology and microstructure of the as-prepared products were characterized by X-ray diffraction, transmission electron microscopy, Fourier transform infrared spectroscopy, X-ray photoelectron spectroscopy and nitrogen adsorption/desorption isotherms. The electrochemical properties of the manganese silicate were evaluated by cyclic voltammetry, galvanostatic charge/discharge and electrochemical impedance spectroscopy. With increasing the hydrothermal time from 0.5 to 3 h, the morphology of the product converted from aggregates composed of nanoparticles into thin drapes with an amorphous and porous structure. After hydrothermal reaction for 3 h, the porous product with a large specific surface area of 439 m2 g−1 exhibits a specific capacitance of 281 F g−1 at a current density of 1 A g−1 in 1 M KOH electrolyte as an electrode material for supercapacitors (SCs). Meanwhile, the product also demonstrates excellent rate performance. Thus the manganese silicate will be an alternative candidate for the electrode material of SCs.
1. Introduction
Supercapacitors (SCs) have attracted intense attention these past few years as one kind of new generation of clean energy-storage systems, which could act as a supplements for batteries, backup power sources, and standby power systems.1 According to the charge storage mechanism, the SCs could be divided into electric double-layer capacitors (EDLCs) and pseudocapacitors.2,3 The energy storage in the EDLCs depends on the formation of an electric double-layer between the electrode surface and electrolyte. Porous carbons are the typical electrode materials for the EDLCs because of their excellent electronic conductivity, low cost, high specific surface area and high chemical stability,4 but their specific capacitance is comparatively low.5 The energy storage in pseudocapacitors is associated with the rapid oxidation–reduction reactions on the electrode surface.6,7 Transition metal nitrides,8,9 tungsten sulfides,10 phosphides11 and vast metal oxides, such as RuO2,12 MnO2,13 NiO,14 VOx,15 Co3O4,16 and Fe2O3,17 have been explored as electrode materials.Among multitudinous active electrode materials, manganese oxides are one kind of the most attractive candidates owing to their low cost, natural abundance and environmental benignity.18–21 MnO2 electrode exhibits two charge storage mechanisms, the insertion and extraction of H+ or alkali metal cations on the MnO2 surface during redox reaction, as well as the adsorption of cations in the electrolyte on the MnO2 electrode.22,23 Nanostructured MnO2 prepared by hydrothermal technique showed a good pseudocapacitive behavior with a specific capacitance of 168 F g−1 for 100 cycles at the current density of 200 mA g−1.22 In order to improve the electric conductivity and charge storage capability of manganese oxides, carbon materials were generally introduced. When 10 wt% Mn2O3 was coated with carbon aerogel microbead, a specific capacitance of 368 F g−1 was achieved at 1 A g−1.24 The graphene/Mn3O4 composites revealed a specific capacitance of 115 F g−1 at 1 A g−1 and the capacitance was almost 100% retained after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at 5 A g−1.25 In addition, manganese hydroxide could also act as electrode material for SCs, and Mn(OH)2 nanoparticles synthesized by sonochemical method displayed a specific capacitance of 127 F g−1 at 0.5 mA cm−2,26 owing to the oxidation from Mn2+ to Mn3+ and then to Mn4+.27,28 It is thus clear that Mn-based materials play an important role in the electrode materials for SCs. Besides the manganese oxides and hydroxide, MnCO3 was very recently reported to be a potential electrode material for SCs,29 which achieved a specific capacitance of 216 F g−1 in 0.1 M Mg(ClO4)2 electrolyte at a current of 0.5 mA and a loading of 1.5 mg cm−2. Meanwhile, MnCO3 also possessed good reversibility, high coulombic efficiency, moderate rate performance and long cycle-life. Therefore, it is of great significance to explore other types of Mn-based materials as the electrode materials for SCs.
000 cycles at 5 A g−1.25 In addition, manganese hydroxide could also act as electrode material for SCs, and Mn(OH)2 nanoparticles synthesized by sonochemical method displayed a specific capacitance of 127 F g−1 at 0.5 mA cm−2,26 owing to the oxidation from Mn2+ to Mn3+ and then to Mn4+.27,28 It is thus clear that Mn-based materials play an important role in the electrode materials for SCs. Besides the manganese oxides and hydroxide, MnCO3 was very recently reported to be a potential electrode material for SCs,29 which achieved a specific capacitance of 216 F g−1 in 0.1 M Mg(ClO4)2 electrolyte at a current of 0.5 mA and a loading of 1.5 mg cm−2. Meanwhile, MnCO3 also possessed good reversibility, high coulombic efficiency, moderate rate performance and long cycle-life. Therefore, it is of great significance to explore other types of Mn-based materials as the electrode materials for SCs.
Mesoporous materials have attracted considerable attention in developing SCs because of their unique pore arrays and large specific surface areas. The mesopores (usually 2–8 nm) could accelerate the kinetic process of ion diffusion in the electrodes and improve the power performance at high current densities.2 Ordered mesoporous carbon materials are representative for SCs, which exhibit excellent rate performance and electric conductivity in ionic electrolyte owing to hierarchically ordered porous structure coupled with large ion-accessible surface area and nitrogen-doping effect.30 However, the limited specific capacitance and high fabrication cost of mesoporous carbon restrain their applications.31
Silicates are safe and low cost materials, which have been widely used in fillers,32 catalysts, adsorption and separation. The basic structural unit of silicates is the tetrahedral anion with four negative charges, which could link to form different structures through various ways. Manganese silicates have been reported to possess mesoporous structure and large specific surface area,33 conducive to ion diffusion and storage. The carbon-coated manganese silicate as anode material for lithium storage exhibited outstanding rate performance and high-rate cycling stability.34 Mesoporous lithium manganese silicate showed excellent reversibility with a capacity of 175 mA h g−1 when cycled in a constant current–constant voltage mode at a current rate of 20 mA g−1.35 However to date, no report has been concerning with the application of porous manganese silicates in SCs.
In the present work, we prepared manganese silicate by the hydrothermal reaction between Na2SiO3·9H2O and MnCl2·4H2O. After hydrothermal reaction for 3 h, the porous product with a large specific surface area of 439 m2 g−1 exhibits a high electrochemical capacitance of about 281 F g−1 at 1 A g−1 in 1 M KOH electrolyte.
2. Experimental section
Analytically pure MnCl2·4H2O, Na2SiO3·9H2O, polyvinylidene fluoride (PVDF), acetylene black, 1-methyl-2-pyrrolidone, Na2SO4 and KOH were purchased from Sinopharm Chemical Reagent. Manganese silicate was synthesized by the simple reaction between Na2SiO3·9H2O and MnCl2·4H2O with a molar ratio of 1:1 according to the following equation.| MnCl2 + Na2SiO3 = MnSiO3 + 2NaCl |
The water solution of 20 mL MnCl2·4H2O with a concentration of 2.5 mol L−1 was titrated into the solution of 50 mL Na2SiO3·9H2O with a concentration of 1 mol L−1 under vigorous magnetic stirring. The uniform mixture was transferred into in a Teflon-lined stainless steel autoclave of 100 mL in capacity and heated at 180 °C. After heating for various hours, the product collected from the autoclave was washed with deionized water 3 times to eliminate the by-product of NaCl, and dried at 105 °C for 12 h. The resultant products obtained from hydrothermally treating for 0.5, 1, 3 and 5 h are designated as S0.5, S1, S3 and S5, respectively.
The products were analyzed by X-ray diffraction (XRD) patterns at a scanning rate of 4° per min in a Rigaku Dmax-2500 diffractometer with Ni filtered Cu Kα radiation (V = 50 kV, I = 100 mA). The morphology was characterized by transmission electron microscopy (TEM) in a JEOL JEM-2100 high-resolution transmission electron microscopy. Infrared (IR) spectrum analysis was carried out in a TENSOR 37 Fourier transform infrared (FTIR) spectroscopy to determine the surface functional groups. The elements in the products were tested by an energy-dispersive X-ray (EDX) spectrometer attached to a field emission scanning electron microscope. The surface elemental composition and oxidation state of the products were obtained by X-ray photoelectron spectra (XPS) on a KRATOS XSAM800 X-ray photoelectron spectrometer (Kratos Analytical Ltd., Manchester, U.K.) using Al Kα radiation (hν = 1486.6 eV) as the excitation source (V = 12 kV, I = 10 mA). The specific surface area and pore structure of the products were acquired by nitrogen adsorption/desorption isotherms in a Quadrasorb SI sorption analyzer at 77 K. The specific surface area was calculated by Brunauer–Emmett–Teller (BET) method, and pore size distribution was determined by Density-Functional-Theory method (DFT) using the adsorption branches of N2 isotherms.
The electrochemical performance was tested in a three-electrode system (calomel electrode as reference electrode, platinum sheet as counter electrode, and the as-prepared products as working electrode) using an IviumStat electrochemistry workstation. The working electrode was fabricated by mixing manganese silicate, 9 wt% PVDF, and carbon black in 1-methyl-2-pyrrolidone with a weight ratio of 8:1:1. The uniform slurry was coated on nickel foam, dried at 120 °C for 12 h in vacuum and pressed at 9 MPa for 30 s. The effective area of active materials on each current collector was about 3 mg cm−2. All the electrochemical performance measurement was tested under air condition.
3. Results and discussion
3.1 Structure and morphology characterization
The XRD patterns of the products are revealed in Fig. 1a. The absence of any distinguishable diffraction peaks in the patterns for S0.5, S1 and S3 indicates that the as-prepared products are in amorphous state. A comparatively strong diffraction peak is observed at 2θ = 33° in the XRD pattern of S5, and the peaks are in good agreement with those of (Mn2O3)3MnSiO3 (JCPDS 74-1206) because the surface Mn2+ is readily oxidized to Mn3+ in water solution. The crystallization degree increases with prolonging the hydrothermal time (Fig. A1 in ESI†), but the product is still (Mn2O3)3MnSiO3 even hydrothermally treating 72 h. After heating S5 in air at 800 °C for 5 h, the product (S5-800) reveals high crystallization degree (Fig. A2 in ESI†), and the diffractions could be assigned to those from MnSiO3 (JCPDS 76-0522) and (Mn2O3)3MnSiO3. The element content in the amorphous products was evaluated by EDX spectrum, and the elements in S3 are depicted in Fig. 1b. The atomic ratio of Mn/Si is close to 1:1 and no other elements are detected except for Mn, Si and O. The EDX spectra for S0.5, S1 and S5 (not presented here) are similar to that for S3.
 | ||
| Fig. 1 (a) XRD patterns of S0.5, S1, S3 and S5, (b) EDX spectrum of S3, and (c) FTIR spectra of S0.5, S1, S3 and S5. | ||
To determine the surface functional groups of the as-prepared manganese silicate, FTIR spectra were measured in the range of 400–4000 cm−1 (Fig. 1c). The peak positions for S0.5, S1, S3 and S5 are approximately coincident. The two strong band around 3369 and 1620 cm−1 are attributed to the O–H vibrations, suggesting that some hydroxyl groups present in the as-prepared manganese silicate despite drying at 105 °C for 12 h.34 The peaks around 1018 and 614 cm−1 are ascribed to the Si–O vibrations in SiO32–36 further confirming the formation of manganese silicate during the hydrothermal treatment.
The surface chemical composition of manganese silicate was further analyzed by XPS (taking S3 as an example due to the similarity of XPS for S0.5, S1 and S5 to those for S3). The survey spectrum of S3 (Fig. 2a) indicates the presence of Si, Mn and O elements in the products, in good agreement with the EDX spectrum in Fig. 1b. The C 1s spectrum around 284.8 eV is attributed to the C–C bond, which could act as a reference to analyze the binding energies of other elements. The high resolution Mn 2p spectrum in Fig. 2b includes two typical peaks around 642.2 (Mn 2p3/2) and 653.6 eV (Mn 2p1/2),37 which could be respectively deconvoluted into three peaks. The peaks at 641.9 and 653.4 eV result from the Mn2+ in MnSiO3,38 those at 642.0 and 653.7 eV from Mn3+,39 and those at 642.6 and 653.9 eV from Mn4+.37 The Mn2+ is prone to be oxidized to Mn3+ and Mn4+ in water solution, and the presence of high valent Mn is advantageous to the pseudocapacitance.40
 | ||
| Fig. 2 XPS of S3 (a) survey spectrum and (b) Mn 2p. | ||
Detailed morphology and structure information of the four products were further obtained by TEM. The TEM images of the products are depicted in Fig. 3. After hydrothermally treating for 0.5 h, a lot of aggregations comprised of nanoparticles were observed in S0.5 (Fig. 3a). When the hydrothermal time was prolonged to 1 h (S1), besides some residual aggregations, there are a lot of thin drapes formed around the aggregations with the creation of a small amount of macropores (Fig. 3b). The macropores are favorable for electrolyte diffusing into the inner micropores. When the hydrothermal time reached 3 h (S3), the aggregations converted almost thoroughly into the drapes with the occurrence of a large number of mesopores and a few macropores (Fig. 3c), conducive to the ion transport. However, when the hydrothermal time is up to 5 h (S5), the drapes linked up into large pieces with the disappearance of macropores (Fig. 3d). From the selected electron diffraction (SED) patterns in the insets, the diffuse halo ring for S0.5, S1 and S3 implies that the products are amorphous, while the weak diffraction rings for S5 denotes the formation of crystalline phase in the product, consistent with the XRD result. The transition from amorphous to crystalline phase is an indication of the densification in structure.
 | ||
| Fig. 3 TEM images of S0.5 (a), S1 (b), S3 (c), and S5 (d). The inset is the corresponding SED pattern. | ||
The porous structure and specific surface area were characterized by nitrogen adsorption–desorption isotherms (Fig. 4). The IV type isotherms with strong N2 adsorption at low pressure and a small hysteresis loop at higher relative pressure of 0.8–1.0 suggest that the products possess microporous and mesoporous structure.41,42 Calculated by the BET method, the specific surface areas are 192 m2 g−1 for S0.5 (Fig. 4a), 270 m2 g−1 for S1 (Fig. 4b), 439 m2 g−1 for S3 (Fig. 4c), and 322 m2 g−1 for S5 (Fig. 4d). The ion-accessible porous structure with large specific surface area is propitious to ion diffusion and storage in the electrode material.43 The pore size distribution was acquired by the adsorption branches of N2 isotherms using the DFT method,44 as depicted in the insets of Fig. 4. A large number of macropores up to 100 nm and some micropores with an average pore size of 1.35 nm are observed in S0.5. With increasing the hydrothermal time, the proportion of micropore and mesopore increases while that of macropore decreases. Small pores peaked at 2.73 nm are founded in S1 verifying the presence of mesopores in the products with the hydrothermal time prolonged to 1 h.44 The continuous pore size distribution of S3 demonstrates the coexistence of micropore, mesopore and macropore in this sample. When the hydrothermal time reached 5 h, micropores and mesopores account for the overwhelming majority. The pore size distribution result agrees well with the TEM result. The pore volumes acquired by the BJH model (cumulative volume of pores between 1.7 and 300.0 nm in diameter) are 0.736 cm3 g−1 for S0.5, 0.789 cm3 g−1 for S1, 0.864 cm3 g−1 for S3 and 0.688 cm3 g−1 for S5, i.e. S3 exhibits the largest pore volume in the samples. The micropores could offer space for ion storage, while the mesopores and macropores are favorable to electrolyte diffusion and ion transport. Thus S3 might possess high specific capacitance and rate capability while used as the electrode material for SCs.
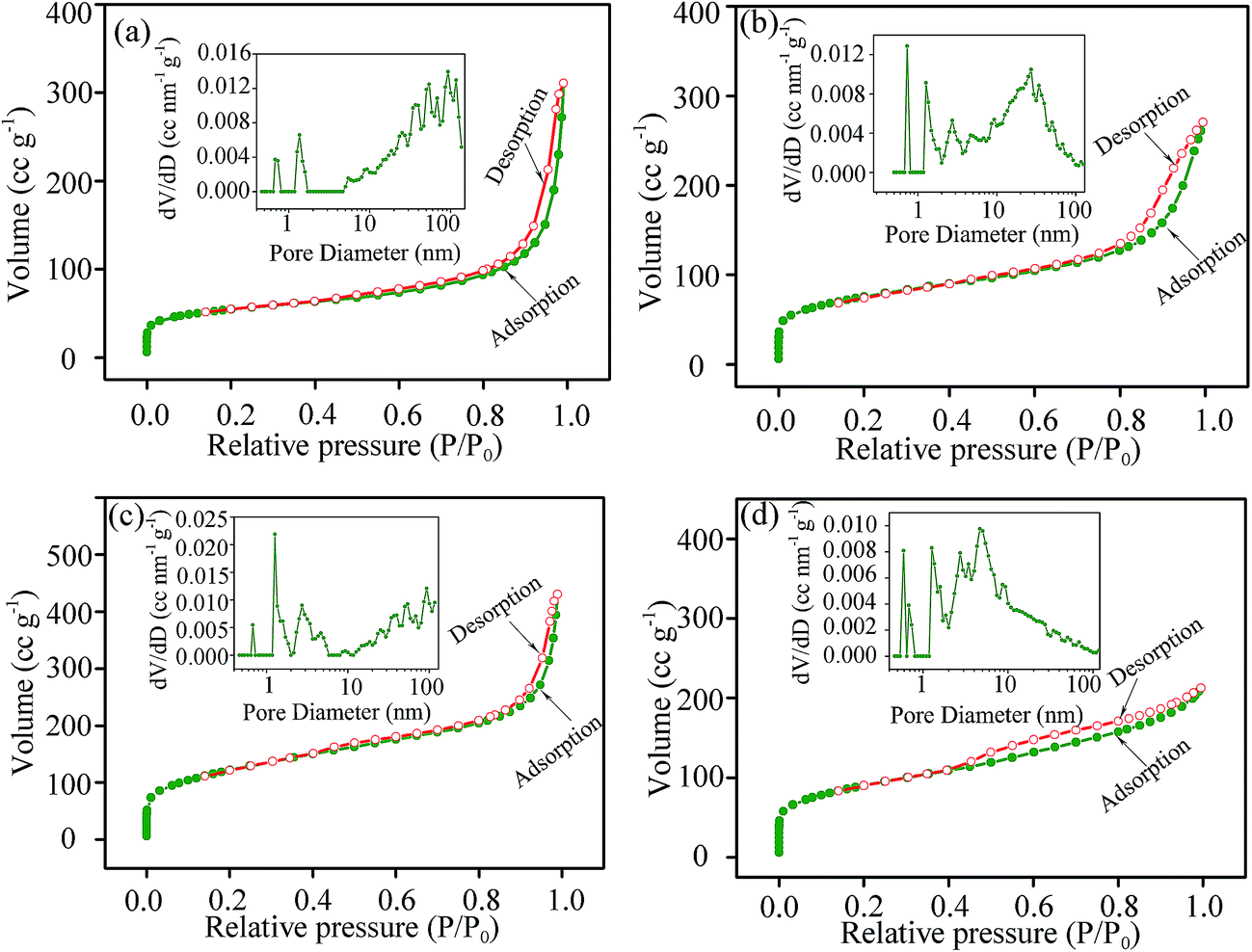 | ||
| Fig. 4 Nitrogen adsorption–desorption isotherms of S0.5 (a), S1 (b), S3 (c) and S5 (d). The inset illustrates the corresponding pore size distribution. | ||
3.2 Electrochemical performance
To evaluate the electrochemical performance of the as-prepared manganese silicate as SC electrodes, cyclic voltammograms (CV) were tested in two different electrolytes of 1 M Na2SO4 and 1 M KOH at a scan rate of 10 mV s−1. The CV plots were recorded after the electrode was firstly stabilized by 100 cycles. From the plots obtained in 1 M Na2SO4 (Fig. 5a), the near-rectangle shape in the potential range of 0.0–0.9 V vs. Hg/Hg2Cl2 suggests that the capacitance results from the EDLC behavior with outstanding reversibility.45 The CV curves achieved in 1 M KOH (Fig. 5b) also reveal symmetrical and near-rectangle shape in the potential range of −0.5–0.4 V vs. Hg/Hg2Cl2. However, a weak electrochemical polarization occurs in the potential range of −0.4–0.0 V for the electrodes, especially in S0.5 and S1. The polarization associates with the marked pseudocapacitive contribution resulted from the rapid Mn4+/Mn3+ redox reactions near the surface of the electrode.46 The specific capacitance could be calculated from the integral area encircled by CV plots in accordance with eqn (1).47
 | (1) |
 | ||
| Fig. 5 CV plots at 10 mV s−1 (a and b) and galvanostatic charge/discharge curves (c and d) of S0.5, S1, S3 and S5 in 1 M Na2SO4 (a and c) and 1 M KOH (b and d). | ||
From eqn (1), the specific capacitance is proportional to the integral area enclosed by CV plots. Apparently, the integral area for S0.5, S1, S3 and S5 in 1 M Na2SO4 is far smaller than that in 1 M KOH at the same scan rate of 10 mV s−1. The specific capacitances calculated by eqn (1) are 165 F g−1 for S0.5 (m = 0.020 g), 208 F g−1 for S1 (m = 0.019 g), 225 F g−1 for S3 (m = 0.016 g) and 206 F g−1 for S5 (m = 0.018 g) in 1 M KOH at the scan rate of 10 mV s−1, and are 72 F g−1 for S0.5, 107 F g−1 for S1 155 F g−1 for S3 and 111 F g−1 for S5 in 1 M Na2SO4 electrolyte, so the manganese silicate electrodes exhibit superior electrochemical performance in 1 M KOH electrolyte to that in 1 M Na2SO4 electrolyte. The larger specific surface areas of S3 (538 m2 g−1) and S5 (558 m2 g−1) than S0.5 (352 m2 g−1) and S1 (434 m2 g−1) provide more ion-accessible surfaces for ion diffusion and storage, so S3 and S5 reveal larger electric double layer capacitance. In addition, the Faraday redox reactions occurred between Mn4+ and Mn3+ on the surface could also contribute to the specific capacitance of manganese silicate.
To acquire further electrochemical information about the manganese silicate electrode in the two different electrolytes, galvanostatic charge–discharge measurement was implemented for S0.5, S1, S3 and S5 at 1 A g−1 (Fig. 5c and d). The charge–discharge curves are nearly linear with negligible voltage drop in both 1 M Na2SO4 and 1 M KOH electrolytes, indicating that the internal resistance of the electrode system is very small.48 The operation voltage was assigned to be 0.9 V by adjusting the potential range to keep the triangular shape of the charge–discharge curves. The specific capacitance could be calculated by the charge/discharge curves according to eqn (2).49
| C = IΔt/(mΔV) | (2) |
By calculation from eqn (2), the specific capacitances are 77 F g−1 for S0.5, 119 F g−1 for S1, 170 F g−1 for S3 and 113 F g−1 for S5 in 1 M Na2SO4 electrolyte (Fig. 6c), and are 173 F g−1 for S0.5, 220 F g−1 for S1, 281 F g−1 for S3 and 226 F g−1 for S5 in 1 M KOH electrolyte (Fig. 6d), consistent with the CV result that the specific capacitance in the 1 M KOH is higher than that in the 1 M Na2SO4 at the same current density of 1 A g−1.
 | ||
| Fig. 6 (a) CV plots at different scan rates and (b) galvanostatic charge/discharge curves at varied current densities of S3 in 1 M KOH electrolyte. | ||
In view of the high capacitance of S3, the CV plots at other scan rates from 20 to 100 mV s−1 were also measured in 1 M KOH electrolyte (Fig. 6a). The specific capacitance decreases gradually with increasing the scan rate. At high scan rates, the migration of ions is not so fast, there are some surfaces of the electrode material that the ions cannot arrive to store charge, and thus the specific capacitance decreases with the high scan rate.47 The CV plot still maintains the near-rectangle appearance even at 100 mV s−1, indicative of the good rate performance of S3.50
In addition, the galvanostatic charge/discharge curves at various current densities were also tested in 1 M KOH electrolyte for S3 (Fig. 6b). The near-linear curves with small deviation demonstrate that the capacitance in the amorphous manganese silicate dominantly results from the capacitive behavior as well as some contributions from pseudocapacitance due to the redox reactions between Mn4+ and Mn3+ on the surface, further confirming the CV results. The specific capacitance is 283 F g−1 at 0.5 A g−1, 281 F g−1 at 1 A g−1, 210 F g−1 at 2 A g−1 and 132 F g−1 at 4 A g−1. The specific capacitance of S3 is pretty stable when the current density is not higher than 1 A g−1, because the mesoporous channels afford efficient pathways for ion diffusion and more effective surface area to store charge. However, the capacitance decreases greatly with increasing the current density to over 2 A g−1 due to the overpotential caused by concentration polarization.51 Compared with the supercapacitor performance of other Mn-based compounds (Table 1), the manganese silicate drapes are good candidate for SC applications.
| Electrode materials | Electrolyte | Current density (A g−1) | Capacitance (F g−1) | Cyclability (F g−1/cycles) |
|---|---|---|---|---|
| MnO2 nanowire/CNT composite paper5 | 0.1 M Na2SO4 | 0.77 | 107.9 | 95/1000 |
| Nanostructured MnO2 (ref. 22) | 1 M Na2SO4 | 0.2 | 168 | 139/100 |
| MnCO3 (ref. 29) | 0.1 M Mg(ClO4)2 | 0.5 mA | 210 | 194/500 |
| Pure Mn3O4 (ref. 52) | 1 M Na2SO4 | 2.0 | 70.0 | 49.8/1500 |
| Graphene/Mn3O4 composite52 | 1 M Na2SO4 | 2.0 | 210.0 | 192/1500 |
| GO–MnO2 nanocomposites53 | 1 M Na2SO4 | 0.2 | 197.2 | 165.9/1000 |
| Birnessite-type MnO2 nanospheres54 | 1 M Na2SO4 | 1.6 | 165 | 160/300 |
| Manganese silicate drapes (this work) | 1 M KOH | 1.0 | 281 | 210/1000 |
As confirmed by the FTIR spectra, hydroxyl groups are the surface functional groups in the as-prepared MnSiO3. To explore the influence of hydroxyl groups on the charge storage mechanism in the electrodes, S1 was further heated at 600 °C for 5 h under nitrogen atmosphere and the product was assigned to S1a. The FTIR spectrum and CV plot of S1a in 1 M KOH electrolyte were also measured (Fig. A3 and A4 in ESI†). Compared with the FTIR spectrum of S1, the content of hydroxyl group decreases significantly after the heat treatment. The calculated specific capacitance of S1a is about 161 F g−1 (m = 0.021 g), lower than that of S1 (208 F g−1). The position of polarization peak for S1a is similar to that for S1, demonstrating the analogous energy storage mechanism despite the marked decrease in hydroxyl group. From the performance comparison, the hydroxyl groups favor to enhance the EDLC performance because they provide excellent wettability with the electrolyte for reducing diffusive resistance to ion transport.50
Power density and energy density are also important indexes for the electrochemical performance of SCs. The power density and energy density could be calculated by the eqn (3) and (4), respectively.50
| E = 0.5CΔV | (3) |
| P = E/Δt | (4) |
The Ragone plot of S3 in 1 M KOH electrolyte is presented in Fig. 7a. The energy density is about 114 W h kg−1 at a power density of 223 W kg−1, and as high as 44 W h kg−1 even at a high power density of 2000 W kg−1. The high energy and power density of manganese silicate drapes are related to the large specific surface area of the mesoporous–microporous structure, because the porous structure is beneficial to increase the contact with electrolyte to enhance the charge storage density.
 | ||
| Fig. 7 (a) Ragone plot and (b) cycling performance of S3 in 1 M KOH at 1 A g−1. | ||
The cycling stability of S3 was assessed in 1 M KOH electrolyte by the galvanostatic charge–discharge at the current density of 1 A g−1 for 1000 cycles (Fig. 7b). In the initial 300 cycles, the capacitance decreased gradually due to manganese dissolution caused by the irreversible oxygen evolution reaction.40 The irreversible reduction of Mn4+ with the manganese dissolution resulted in the decrease of active materials on the electrode, giving rise to a loss of pseudocapacitance. Additionally, the slight corrosion occurring at the electrode/current collector interface in the charge/discharge process led to the increase of the equivalent series resistance in the electrode system.40 After 300 cycles, the specific capacitance tended to be stable, and a specific capacitance of 210 F g−1 was still retained after 1000 cycles, higher than that of MnCO3 (194 F g−1 after 500 cycles29).
In order to further understand the charge transport kinetics of manganese silicate drapes, electrochemical impedance spectra (EIS) were measured in the frequency range between 100 kHz and 0.01 Hz by applying a 10 mV amplitude signal (Fig. 8). The Nyquist plots consist of an inclined line at low frequency and a semicircle at high frequency.55 The intercept of the semicircle (Re) represents the electrolyte resistance, and the semicircle diameter (Rct) denotes the charge-transfer resistance.56 In Fig. 8, the inclined line in the low frequency region deflects to Zim axis, indicating the electrode materials have good capacitive behavior and the electrolyte could easily diffuse to the surface. The charge-transfer resistance is 5 Ω for S0.5 (Fig. 8a), 4 Ω for S1 (Fig. 8b) 4.5 Ω for S5 (Fig. 8d) and only about 0.5 Ω for S3 (Fig. 8c). A large number of mesopores and macropores with high surface area for the drapes in S3 increases the contact area between the electrode materials and electrolyte, thus decreasing the charge-transfer resistance and enhancing the performance.
 | ||
| Fig. 8 EIS of S0.5 (a), S1 (b), S3 (c) and S5 (d). | ||
From the above analysis, the good electrochemical performance of the manganese silicate drapes as electrode material for SCs is relevant to the following aspects. (1) After hydrothermally treating for 3 h, the manganese silicate drapes possess high specific surface area (439 m2 g−1) with numerous micropores, advantageous to increasing the contact with electrolyte and enhancing the charge storage density. (2) The manganese silicate drapes have a large number of mesopores and macropores, effectively reducing the transport distance of electrolyte ion in the charge/discharge process. (3) The existence of hydroxyl groups could improve the hydrophilicity of manganese silicate drapes, conducive to reducing the ion transfer resistance. (4) Mn-ions could transform between different valences, contributing to the pseudocapacitive performance. Therefore, the capacitance of manganese silicate drapes is dominated by an EDLC behavior and also by pseudocapacitive behavior to some degree.
4. Conclusions
In summary, manganese silicate drapes with porous structure were synthesized by the hydrothermal reaction between Na2SiO3·9H2O and MnCl2·4H2O. When used as electrode material for SCs, the product obtained by hydrothermally treating 3 h exhibits a specific capacitance of 281 F g−1 at 1 A g−1 and also shows good rate performance in 1 M KOH. In brief, the manganese silicate is a promising electrode material for SCs, and this investigation will impel the exploration of metal silicates for SC applications.Acknowledgements
This work was supported by Shandong Provincial Natural Science Foundation, PR China (ZR2015EM016), Science and Technology Development Project of Shandong Province, PR China (2016GGX102031 and 2015GGX102005).References
- J. R. Miller and P. Simon, Science, 2008, 321, 651–652 CrossRef CAS PubMed.
- L. L. Zhang and X. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- E. Frackowiak, Phys. Chem. Chem. Phys., 2007, 9, 1774–1785 RSC.
- S.-L. Chou, J.-Z. Wang, S.-Y. Chew, H.-K. Liu and S.-X. Dou, Electrochem. Commun., 2008, 10, 1724–1727 CrossRef CAS.
- Y. Huang, J. Liang and Y. Chen, Small, 2012, 8, 1805–1834 CrossRef CAS PubMed.
- B. Li, M. Zheng, H. Xue and H. Pang, Inorg. Chem. Front., 2016, 3, 175–202 RSC.
- T. Zhai, X. Lu, Y. Ling, M. Yu, G. Wang, T. Liu, C. Liang, Y. Tong and Y. Li, Adv. Mater., 2014, 26, 5869–5875 CrossRef CAS PubMed.
- W. Li, C.-Y. Cao, C.-Q. Chen, Y. Zhao, W.-G. Song and L. Jiang, Chem. Commun., 2011, 47, 3619–3621 RSC.
- S. Ratha and C. S. Rout, ACS Appl. Mater. Interfaces, 2013, 5, 11427–11433 CAS.
- X. Wang, W. Li, D. Xiong and L. Liu, J. Mater. Chem. A, 2016, 4, 5639–5646 CAS.
- B. Li, M. Zheng, H. Xue and H. Pang, Inorg. Chem. Front., 2016, 3, 175–202 RSC.
- M.-W. Xu, D.-D. Zhao, S.-J. Bao and H.-L. Li, J. Solid State Electrochem., 2007, 11, 1101–1107 CrossRef CAS.
- K. C. Liu and M. A. Anderson, J. Electrochem. Soc., 1996, 143, 124–130 CrossRef CAS.
- Y. Yan, B. Li, W. Guo, H. Pang and H. Xue, J. Power Sources, 2016, 329, 148–169 CrossRef CAS.
- S. K. Meher and G. R. Rao, J. Phys. Chem. C, 2011, 115, 15646–15654 CAS.
- D. Wang, Y. Li, Q. Wang and T. Wang, J. Solid State Electrochem., 2012, 16, 2095–2102 CrossRef CAS.
- W. Wei, X. Cui, W. Chen and D. G. Ivey, Chem. Soc. Rev., 2011, 40, 1697–1721 RSC.
- Z. Yu, B. Duong, D. Abbitt and J. Thomas, Adv. Mater., 2013, 25, 3302–3306 CrossRef CAS PubMed.
- X. Lu, T. Zhai, X. Zhang, Y. Shen, L. Yuan, B. Hu, L. Gong, J. Chen, Y. Gao and J. Zhou, Adv. Mater., 2012, 24, 938–944 CrossRef CAS PubMed.
- P. Yang, Y. Ding, Z. Lin, Z. Chen, Y. Li, P. Qiang, M. Ebrahimi, W. Mai, C. P. Wong and Z. L. Wang, Nano Lett., 2014, 14, 731–736 CrossRef CAS PubMed.
- V. Subramanian, H. Zhu and B. Wei, J. Power Sources, 2006, 159, 361–364 CrossRef CAS.
- R. K. Sharma, H.-S. Oh, Y.-G. Shul and H. Kim, J. Power Sources, 2007, 173, 1024–1028 CrossRef CAS.
- X. Wang, L. Liu, X. Wang, L. Yi, C. Hu and X. Zhang, Mater. Sci. Eng., B, 2011, 176, 1232–1238 CrossRef CAS.
- J. W. Lee, A. S. Hall, J.-D. Kim and T. E. Mallouk, Chem. Mater., 2012, 24, 1158–1164 CrossRef CAS.
- S. Anandan, B. G. S. Raj, G.-J. Lee and J. J. Wu, Mater. Res. Bull., 2013, 48, 3357–3361 CrossRef CAS.
- W.-H. Kao and V. Weibel, J. Appl. Electrochem., 1992, 22, 21–27 CrossRef CAS.
- P. K. Nayak and N. Munichandraiah, Electrochem. Solid-State Lett., 2009, 12, A115–A119 CrossRef CAS.
- S. Devaraj, H. Liu and P. Balaya, J. Mater. Chem. A, 2014, 2, 4276–4281 CAS.
- D. Liu, C. Zeng, D. Qu, H. Tang, Y. Li, B.-L. Su and D. Qu, J. Power Sources, 2016, 321, 143–154 CrossRef CAS.
- K. Xie, X. Qin, X. Wang, Y. Wang, H. Tao, Q. Wu, L. Yang and Z. Hu, Adv. Mater., 2012, 24, 347–352 CrossRef CAS PubMed.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS PubMed.
- J. Qu, W. Li, C.-Y. Cao, X.-J. Yin, L. Zhao, J. Bai, Z. Qin and W.-G. Song, J. Mater. Chem., 2012, 22, 17222–17226 RSC.
- Y.-Y. Wang, T. Li, Y.-X. Qi, R.-L. Bai, L.-W. Yin, H. Li, N. Lun and Y.-J. Bai, Electrochim. Acta, 2015, 186, 572–578 CrossRef CAS.
- R. J. Gummow and Y. He, RSC Adv., 2014, 4, 11580–11584 RSC.
- M. Drygas, J. F. Janik, M. M. Bucko, J. Gosk and A. Twardowski, RSC Adv., 2015, 5, 37298–37313 RSC.
- Z. Lei, J. Zhang and X. Zhao, J. Mater. Chem., 2012, 22, 153–160 RSC.
- A. Grosvenor, E. Bellhouse, A. Korinek, M. Bugnet and J. McDermid, Appl. Surf. Sci., 2016, 379, 242–248 CrossRef CAS.
- B. J. Tan, K. J. Klabunde and P. M. Sherwood, J. Am. Chem. Soc., 1991, 113, 855–861 CrossRef CAS.
- D. Bélanger, L. Brousse and J. W. Long, Electrochem. Soc. Interface, 2008, 17, 49 Search PubMed.
- L. Cao, D. Chen, W. Li and R. A. Caruso, ACS Appl. Mater. Interfaces, 2014, 6, 13129–13137 CAS.
- D. Pérez-Quintanilla, A. Sánchez and I. Sierra, J. Colloid Interface Sci., 2016, 472, 126–134 CrossRef PubMed.
- L.-x. Li and L. Feng, New Carbon Mater., 2011, 26, 224–228 CrossRef CAS.
- S. Xin, L. Gu, N.-H. Zhao, Y.-X. Yin, L.-J. Zhou, Y.-G. Guo and L.-J. Wan, J. Am. Chem. Soc., 2012, 134, 18510–18513 CrossRef CAS PubMed.
- J.-K. Chang, C.-T. Lin and W.-T. Tsai, Electrochem. Commun., 2004, 6, 666–671 CrossRef CAS.
- M. Nakayama, T. Kanaya and R. Inoue, Electrochem. Commun., 2007, 9, 1154–1158 CrossRef CAS.
- H. Li, M. Yu, F. Wang, P. Liu, Y. Liang, J. Xiao, C. Wang, Y. Tong and G. Yang, Nat. Commun., 2013, 4, 1894 CrossRef CAS PubMed.
- W. Zhao, S. Wang, C. Wang, S. Wu, W. Xu and M. Zou, Nanoscale, 2016, 8, 626–633 RSC.
- X.-f. Wang, D.-b. Ruan and Y. Zheng, Trans. Nonferrous Met. Soc. China, 2006, 16, 1129–1134 CrossRef CAS.
- Q. Liang, L. Ye, Z.-H. Huang, Q. Xu, Y. Bai, F. Kang and Q.-H. Yang, Nanoscale, 2014, 6, 13831–13837 RSC.
- W.-H. Jin, G.-T. Cao and J.-Y. Sun, J. Power Sources, 2008, 175, 686–691 CrossRef CAS.
- G. Jin, X. Xiao, S. Li, K. Zhao, Y. Wu, D. Sun and F. Wang, Electrochim. Acta, 2015, 178, 689–698 CrossRef CAS.
- S. Chen, J. Zhu, X. Wu, Q. Han and X. Wang, ACS Nano, 2010, 4, 2822–2830 CrossRef CAS PubMed.
- B. Ming, J. Li, F. Kang, G. Pang, Y. Zhang, L. Chen, J. Xu and X. Wang, J. Power Sources, 2012, 198, 428–431 CrossRef CAS.
- X. Li, Y. Feng, M. Li, W. Li, H. Wei and D. Song, Adv. Funct. Mater., 2015, 25, 6858–6866 CrossRef CAS.
- X. Li, G. Li, C. Fu, D. Luo, J. Fan, D. Xie and L. Li, Technology, 2015, 4, 14 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra19102a |
| This journal is © The Royal Society of Chemistry 2016 |