
Versatile surface charge-mediated anti-fouling UF/MF membrane comprising charged hyperbranched polyglycerols (HPGs) and PVDF membranes†
Abstract
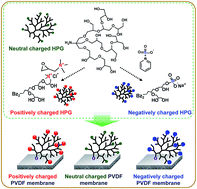
We develop a charge-modified PVDF UF/MF membrane that restricts membrane fouling derived from charged water contaminants. The charge-modified membranes are fabricated through surface assembly of various charged hyperbranched polyglycerols (HPGs), which act as anti-fouling agents. The native HPGs are neutrally charged polymers with a lot of hydroxyl end groups that can be modified with specific charges. We prepare three types of charged HPGs, i.e., neutrally charged HPGs without end group modifications, positively charged HPG with quaternary ammonium groups, and negatively charged HPG with sulfonate groups. The combined results for 1H NMR, FT-IR, and XPS results show that the charged HPGs are successfully synthesized and bound to the surfaces of the PVDF membrane. The surface hydrophilicity improves upon assembly of the hydrophilic charged HPGs. As expected, zeta potential results disclose that the differently charged HPGs provide the desired electrical properties to the membrane surface. In addition, the surface-assembled charged HPGs effectively suppress the attachment and accumulation of foulants having the same charge due to electrostatic repulsion and improved surface hydrophilicity.
 Please wait while we load your content...
Please wait while we load your content...

