
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of catalyst carbon supports on the oxygen reduction reaction in alkaline media: a comparative study
Miguel A. Molina-García and
Neil V. Rees*
Centre for Hydrogen and Fuel Cell Research, School of Chemical Engineering, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: n.rees@bham.ac.uk; Fax: +44 (0)121 414 5324; Tel: +44 (0)121 414 5325
First published on 30th September 2016
Abstract
Some carbon materials commonly used as electronically-conductive supports for catalysts in fuel cell research are Carbon Black (CB), multi-walled carbon nanotubes (MWCNT), Graphene Oxide (GO) and reduced graphene oxide (rGO). Here we present a comparative study into the relative effects of each of these on the performance towards the oxygen reduction reaction (ORR) in alkaline media. For the purposes of comparing the supports, a simple Pt catalyst is used and the performance is evaluated via Koutecky–Levich analysis and direct measurement of peroxide by rotating ring-disk electrode (RRDE) to determine the number of electrons (n) transferred in the ORR. It is found that Pt/CB follows a quasi 4-electron mechanism due to that the ORR takes place mainly on the active Pt particles, whereas Pt/MWCNT, Pt/GO and Pt/rGO exhibit a mixed behaviour between the two proposed mechanisms due to the higher activity of the graphene-derived supports towards the peroxide formation compared to CB. The effect of the oxide groups of GO and the metal impurities of MWCNT on the catalytic performance is also studied.
1. Introduction
Carbon has been widely used as material for catalyst support for several reasons, such as its abundance, potential low cost, availability in a variety of forms with different properties, excellent electronic conductivity, high surface area and relatively inert surface.1 Among the different carbon supports carbon black (CB) is the most widespread and the most common commercial CB support is Vulcan XC-72, consisting of quasi-spherical carbon particles with diameters < 50 nm and polycrystalline structures of several layers with an interplanar separation of 0.35–0.38 nm, reaching a high surface area of around 250 m2 g−1.2 Countless studies investigating CB-supported catalysts for the oxygen reduction reaction (ORR) in alkaline media exist, including many concerning platinum group metal (PGM) catalysts namely Pt/C,3,4 PdO/C,5 Pd–Ag/C6 and even a complete integrated study investigating all the PGM (excluding Os) supported on CB: Pt/C, Pd/C, Rh/C, Ru/C and Ir/C, showing Pd/C and Pt/C the best performance in this experiment.7 CB has been also tested as carbon support in non-PGM catalysts, such as CoFeN/C,8 Au/C,9 Ag/C,9–11 Co-Ppy/C,12 FePc/PANI/C13 or MnOOH/C.14The behaviour of CB as a catalyst support depends strongly on its specific characteristics, as well as on the application of different preparatory treatments. For example, oxidation of CB provides surface groups on the carbon support that facilitate the deposition of Pt.4 In another study, an impregnation/reduction method using the weak reducing agent Na2S2O4 followed by a thermal treatment at 700 °C in Ar produced growth and then crystallisation of Pt particles on the carbon structure.15 Another investigation reported that Vulcan XC-72 treated with citric acid induced the deposition of smaller Pt particles.16 Regarding the physical properties, the specific surface area of the carbon support has been proven to be a crucial factor in the catalytic activity,17 whereas the porosity of the CB support has also been shown to play a role in the activity of a Pt–Ru catalyst towards methanol oxidation.18
Another carbon material increasingly finding application as a catalyst support are carbon nanotubes in either single-walled (SWCNT) or the most common multi-walled (MWCNT) forms. The latter have been shown to display excellent electrocatalytic activity towards ORR in alkaline media.19 MWCNTs have been used as carbon supports for a variety of metal catalysts in a number of studies, including the demonstration that the ORR on Ag nanoparticles deposited on MWCNT showed high electrocatalytic activity towards the peroxide reduction.20 Yu and Manthiram proved that MnNiCoO4 nanoparticles deposited on N-doped MWCNTs have better catalytic selectivity towards ORR than Pt in alkaline media.21 Similarly, experiments to evaluate the performance of a PdNi/MWCNT catalyst towards the methanol oxidation in alkaline media suggest that the electrocatalytic activity of this system is higher than Pd/MWCNT due to a decrease on the reaction overpotential.22 Moreover, the catalytic performance of MWCNT can be modified by chemical functionalization of the nanotubes. Dumitru and co-workers demonstrated that the ORR in alkaline media proceeds by a two-electron mechanism when MWCNT are functionalized with citric acid, whereas it undergoes a four-electron mechanism when the nanotubes are treated with peroxymonosulfuric acid, due to an increase in the number of C–O groups.23
Recent developments on catalyst supports have been made on graphene derivatives, driven by its unique properties such as very high electronic and thermal conductivity, extremely high surface area (theoretically 2630 m2 g−1) and high mechanical strength.24 Although graphene has been proposed as catalyst support itself,25 it cannot be produced for large-scale by micromechanical exfoliation from graphite.26 The present study focuses on two graphene derivatives: graphene oxide (GO) and reduced-graphene oxide (rGO), which can be easily obtained from abundant graphite crystal using well-known oxidation methods,27 followed by a reduction in the case of rGO.28 The main characteristic of GO with respect to graphene is the loss of electronic conductivity due to the partial transition from sp2 to sp3 carbon atoms.29 Several techniques have been reported in the literature to characterise GO, such as X-ray photoelectron spectroscopy (XPS),26 Fourier-transform infrared spectroscopy (FTIR),30 X-ray diffraction (XRD), thermogravimetric analysis (TGA)27 and cyclic voltammetry analysis of colloidal suspensions of GO.31 The reduction of GO would recover part of the characteristic conductivity of graphene by partially restoring the π-network.32 Moreover, the elimination of oxide groups would create vacancies along the sp2 carbon structure which could favour the deposition and dispersion of the catalyst particles.33 GO reduction can be achieved via thermal, electrochemical or chemical methods.34 Of the latter, hydrazine is a selective reagent to reduce GO,28 but has the disadvantages of toxicity and the aggregation of rGO layers leading to the increasing use of L-ascorbic acid as a more benign reductant.35
To the best of our knowledge no integral studies comparing CB, MWCNT, GO and rGO as catalyst carbon supports for the ORR in alkaline media have been carried out to date. In this study we will first characterise the fabricated catalyst-supports via TGA, X-ray fluorescence (XRF), transmission electron microscopy (TEM), FTIR and XPS. Then, the different catalysts will be investigated in ex situ rotating-ring disk electrode (RRDE) experiments by deposition onto a glassy carbon (GC) electrode to study the different kinetic effects of the four carbon supports. AC impedance measurements have been also performed.
2. Experimental procedure and methods
2.1. Preparation of the catalysts and reduction of GO
The synthesis of Pt/C catalysts by a chemical reduction process with sodium borohydride (NaBH4-CRP) followed the procedure of Fang and co-workers.36 195.2 mg of carbon support were deposited into 200 mL of ultrapure water. After 30 minutes of sonication, 5 mL of 0.05 M K2PtCl6 aqueous solution (121.5 mg of K2PtCl6 in 5 mL ultrapure water) was added and stirred at 500 rpm for 3 hours. Next, KOH was added to adjust the pH at 8 and then 5 mL of an aqueous solution containing 94.6 mg of NaBH4 (the amount was calculated to provide a NaBH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pt molar ratio of 10:1) was added to the solution. After stirring at 500 rpm overnight the solution was filtered, thoroughly washing the catalyst with ultrapure water. Finally, the catalyst was left to dry in the oven at 80 °C overnight and then weighed.
:Pt molar ratio of 10:1) was added to the solution. After stirring at 500 rpm overnight the solution was filtered, thoroughly washing the catalyst with ultrapure water. Finally, the catalyst was left to dry in the oven at 80 °C overnight and then weighed.
Carbon black was Ensaco Conductive Carbon Black provided by TIMCAL Graphite & Carbon (a member of IMERYS). MWCNT was provided by Nanostructured & Amorphous Materials INC (Los Alamos, NM 87544, USA), having the next properties: purity: 95%, length: 10–20 μm, diameter: 20–40 nm, SSA > 360 m2 g−1. The preparation of rGO was partially based on the procedure described by Zhang and co-workers.37 For this, 300 mg of commercial GO (Nanoinnova, ref.: GO.M.10.29-1) were deposited into 300 mL of ultrapure water together with 3 g of L-ascorbic acid at room temperature (ca. 298 K). The mixture was left to stir overnight at 500 rpm and then filtered 3 times, washing with copious amount of ultrapure water between filtrations. The catalyst was then dried in the oven at 80 °C overnight and finally weighed to check the amount of rGO obtained.
2.2. Characterisation methods
TGA was carried out using a TG 209 F1 Libra thermo-gravimetric analyser (TA Instruments). The samples were placed in an alumina crucible and then heated from 25 to 900 °C with a heating rate of 10 °C min−1 under a flow of air at 50 mL min−1. The different catalysts were also imaged by a TEM using a JEOL 2100F at 200 kV, equipped with a FEG source and a Gatan Orius CCD camera in order to obtain images with 0.025 nm pixel resolution. XRF was performed using a Niton XL3T TYPE 980 portable XRF analyser + Helium GOLDD for light element analysis. An X-ray tube with a 50 kV Ag anode was used for precious metal spectra evaluation. Catalysts based on GO and rGO carbon supports were characterised by FTIR using a PerkinElmer FT-IR Spectrum 100. XPS were obtained at the National EPSRC XPS Users' Service (NEXUS) at Newcastle University using a Thermo Scientific K-Alpha XPS instrument with a monochromatic Al Kα X-ray source.For the ex situ characterisation of the catalysts rotating ring-disk voltammetry was performed using a Metrohm AutoLAB PGSTAT128N potentiostat in a Faraday cage. The reference electrode was an Ag/AgCl (3 mol dm−3) electrode (E0 = +0.210 V vs. SHE) and the counter electrode was a Pt mesh. The RRDE (E7R9 Series, Pine Instruments Inc., USA) was formed by a GC disk (5.61 mm diameter) and a Pt ring with an area of 0.1866 cm2, with a collection efficiency of 37%. Prior to each experiment the RRDE was thoroughly polished with consecutive alumina slurries of 1, 0.3 and 0.05 μm and then sonicated to remove any impurities. All the experiments were carried out in 0.1 M KOH solution made with ultrapure water (resistivity ≥ 18.2 MΩ cm, milli-Q Millipore), except for electrochemical surface area (ECSA) characterisation carried out in 0.1 M HClO4 solution.
Several catalyst inks were prepared with the different catalysts. The ink was made by mixing 4 mg of the required catalyst with 0.8 mL of isopropyl alcohol (IPA), 3.18 mL of ultrapure water and 0.02 mL of the commercial I2 ionomer solution (Acta S.p.a.) to act as a binder, obtaining an ink with 1 mg mL−1 of catalyst concentration and 0.02% of I2 ionomer. This mixture was then sonicated for 1 hour and after this a 5 μL aliquot was pipetted onto the GC disk. This amount was calculated from the optimal loading of 20 μg cm−2, since for higher loadings the thickness of catalyst film does not satisfy the mass transport characteristics of RRDE.38 After the ink deposition the droplet was left to dry at room temperature for 30 minutes at 700 rpm as described in literature39 in order to get an uniform layer. In the case of ECSA determination, the ink was formed by 5 mg of the catalyst, 1.6 mL of ultrapure water, 0.4 mL of IPA and 20 μL of 5% Nafion solution (Fluka 117-Nafion Solution in a mixture of water and aliphatic alcohols), with a final concentration of 2.5 mg catalyst per mL (1 hour sonication). 10 μL of this ink were pipetted onto a GC electrode (5 mm diameter) and let dry for 30 min at 700 rpm.
The RRDE was then immersed in the alkaline solution and cycled between −1.2 and +1.0 V at 100 mV s−1 until a stable response was observed. The solution was then thoroughly purged with N2 (BOC gases, O2 free, 99.998% purity) and voltammograms were measured to determine the capacitive current and ohmic resistance of the solution. The solution was then bubbled with O2 (BOC gases, N5, 99.999% purity) for 45 minutes until saturation. Linear sweep voltammograms (LSV) were then recorded at 25 mV s−1 between −0.4 and +1.0 V at the rotation speeds from 400 to 2400 rpm. The Pt ring voltage was fixed at +0.47 V to ensure complete HO2− decomposition.40 For the ECSA experiment, the solution was purged with N2 for 30 minutes and cycled between −0.2 V and +0.8 V at 20 mV s−1. The measurements were carried out at 293 ± 2 K.
AC impedance measurements were also taken in potentiostatic mode at 0 V vs. the same Ag/AgCl (3 mol dm−3) ref. electrode in 0.1 M KOH solution purged with N2, with the frequency varying from 100 to 0.01 kHz. The volume of the catalyst solution deposited onto the GC electrode was in this case 6 μL for all the samples, with the rest of the procedure remaining the same as previously explained. The resistance of the layers were not found to depend on the dropcast volume (and hence thickness) in the range from 3 to 12 μL.
3. Results and discussion
3.1. Characterisation of catalyst-supports
TGA analysis was conducted to confirm the actual amount of Pt deposited onto the carbon supports (see Fig. 1). CB and MWCNT display a similar pattern with no significant amount of water and gradual combustion of carbon. For the CB the onset of carbon oxidation (Tonset) is 684 °C compared to MWCNT where Tonset is 484 °C. The residual mass for MWCNT before Pt deposition is 4.5% due to impurities remaining from the fabrication process of nanotubes as reported elsewhere.41 The behaviour of GO and rGO is different due to the presence of oxide groups. In GO there is an initial weight loss of 14.9% up to 150 °C, due to evaporation of water trapped in the GO bulk, followed by a decrease of 27.1% up to 260 °C due to decarboxylation processes. At this point a distortion appears in the graph due to “micro-explosive” decompositions previously reported in literature,42 with final combustion of carbon starting at 470 °C. A similar pattern is observed in rGO, with differences in the degree of decarboxylation being due to the lower level of oxide species compared to GO. Table 1 shows the amount of Pt in the different catalysts calculated from the values of residual mass and the analysis by XRF of the metal residue observed in Fig. 1. The Pt loading can be considered the same for Pt/CB, Pt/MWCNT and Pt/rGO with an average value of (14.9 ± 1.1)%, whereas it is meaningfully higher for Pt/GO with a value of 19.1%, probably due to the effect of the oxide groups during the Pt deposition.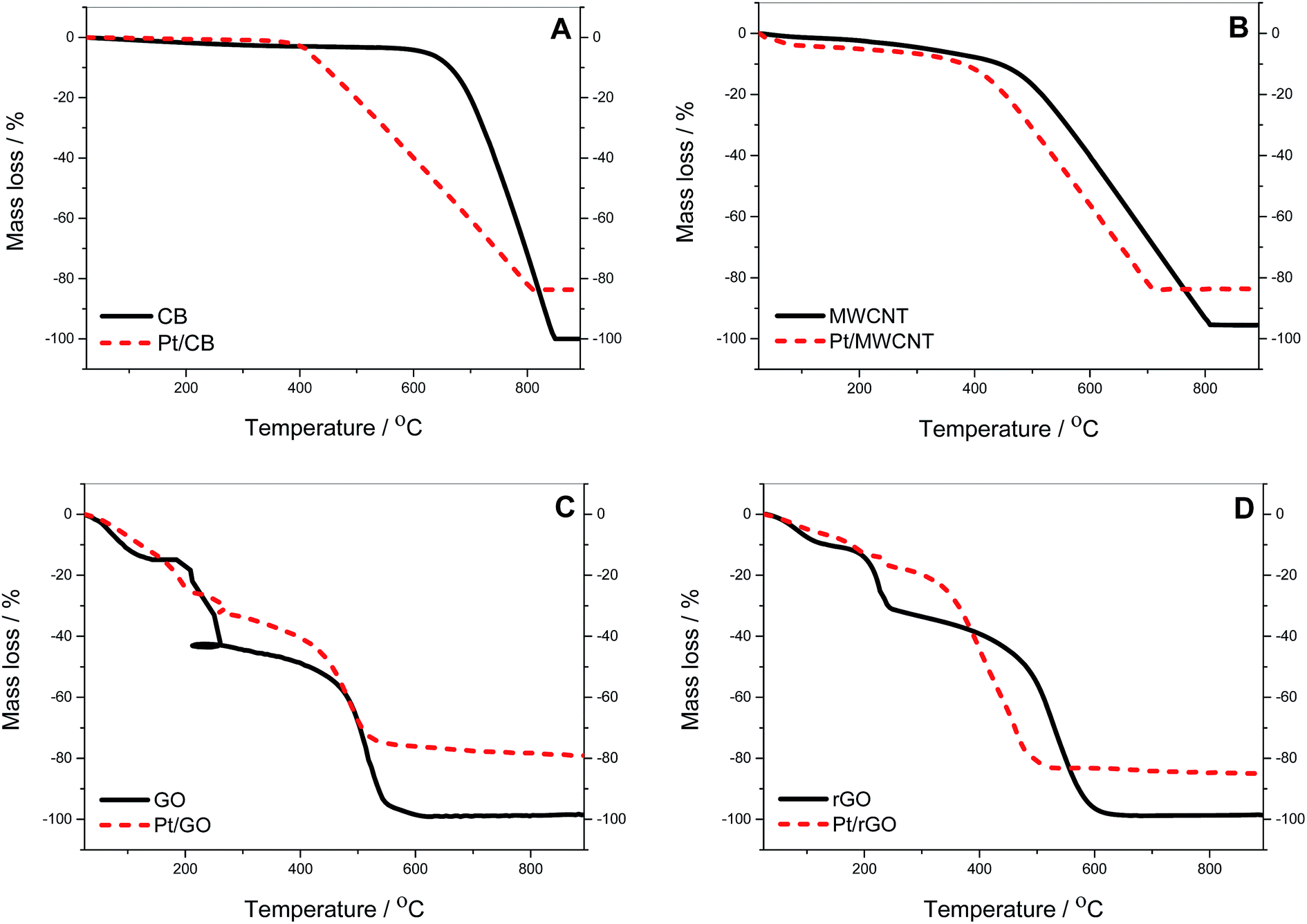 | ||
| Fig. 1 Thermogravimetric mass loss profiles in air at heating rate of 10 K min−1: (A) CB, (B) MWCNT, (C) GO and (D) rGO, before (solid line) and after Pt deposition by NaBH4-CRP (dashed line). | ||
| Pt/CB | Pt/MWCNT | Pt/GO | Pt/rGO | |
|---|---|---|---|---|
| Residual mass (TGA)/% | 16.28 | 16.37 | 20.80 | 14.92 |
| Pt in residue (XRF)/% | 97.54 | 91.26 | 91.83 | 92.38 |
| Pt loading/% | 15.9 | 14.9 | 19.1 | 13.8 |
Fig. 2 shows the TEM images of the different catalysts together with their corresponding particle size distribution. It is remarkable the homogeneity of the particle size in the Pt/CB catalyst, with 80% of the particles being in the range between 3–4 nm and a mean particle diameter (dp) of 3.7 nm. On the contrary, Pt/MWCNT shows very high dispersion of particle size with particles up to 7 nm of diameter, with a value of dp of 4.5 nm. This observation highlights the importance of functionalization of nanotubes, for example by a chemical modification with H2SO4 and/or HNO3, to create nucleation sites which favour a more homogenous Pt distribution.43 The TEM images for Pt/GO and Pt/rGO show a homogeneous particle size in both catalysts with identical value of dp: 3.8 nm. Nevertheless, it can be observed a higher particle agglomeration effect in the case of Pt/GO with respect to Pt/rGO. This may be related to the vacancies created during the reduction of GO with L-ascorbic acid when the oxide groups leave the graphene structure. These have been reported to create binding sites to anchor the Pt precursor ions which are subsequently reduced to Pt particles,44 reducing by this way the particle agglomeration. This good dispersion of Pt particles on rGO structures has been already reported in previous investigations.45–47 As we have seen, the Pt particle diameters are in the range between 2 and 7 nm for all the catalysts. According to some investigations, this range is beyond the particle size that can influence the catalytic behaviour,48 especially regarding the mass activity49 and the ORR performance.50
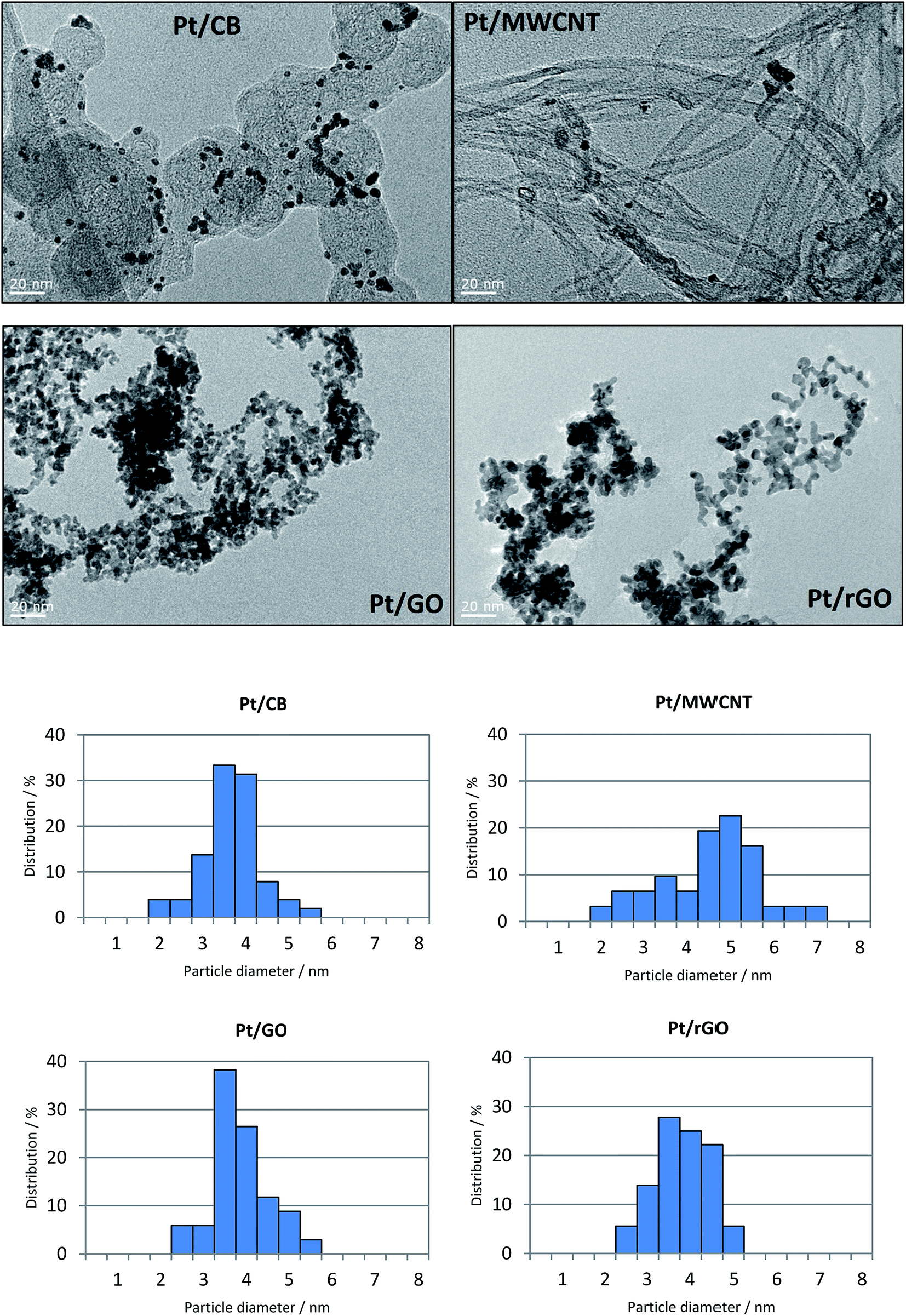 | ||
| Fig. 2 FEG-TEM images showing Pt/CB, Pt/MWCNT, Pt/GO and Pt/rGO catalysts, with their corresponding particle size distribution. | ||
FTIR spectra were also taken to evaluate the influence of the reductive treatments on the oxide groups present in the GO-derived carbon supports (Fig. 3A and B). First, the attenuation of peaks in the characteristic spectra of oxygen-containing moieties (Table 2) in the case of rGO, Pt/GO and Pt/rGO indicates the extent of reduction of the support that has occurred. The peak at 1614 cm−1 in GO supports Montes-Morán and co-workers,51 who assign this peak to cyclic ethers or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching related to carbon rings decorated with phenolic groups rather than to aromatic ring vibrations. Dékány and co-workers52 discovered that a drying process modifies the intensity of this peak, suggesting that it could be related to water present in the GO structure. Another study independently confirms that the peak in this region is related to vibrations of adsorbed water molecules.53 In our case, this could explain why this peak does not appear in the samples that were subjected to a thorough dry procedure as part of the processes for GO reduction and/or Pt deposition.
C stretching related to carbon rings decorated with phenolic groups rather than to aromatic ring vibrations. Dékány and co-workers52 discovered that a drying process modifies the intensity of this peak, suggesting that it could be related to water present in the GO structure. Another study independently confirms that the peak in this region is related to vibrations of adsorbed water molecules.53 In our case, this could explain why this peak does not appear in the samples that were subjected to a thorough dry procedure as part of the processes for GO reduction and/or Pt deposition.
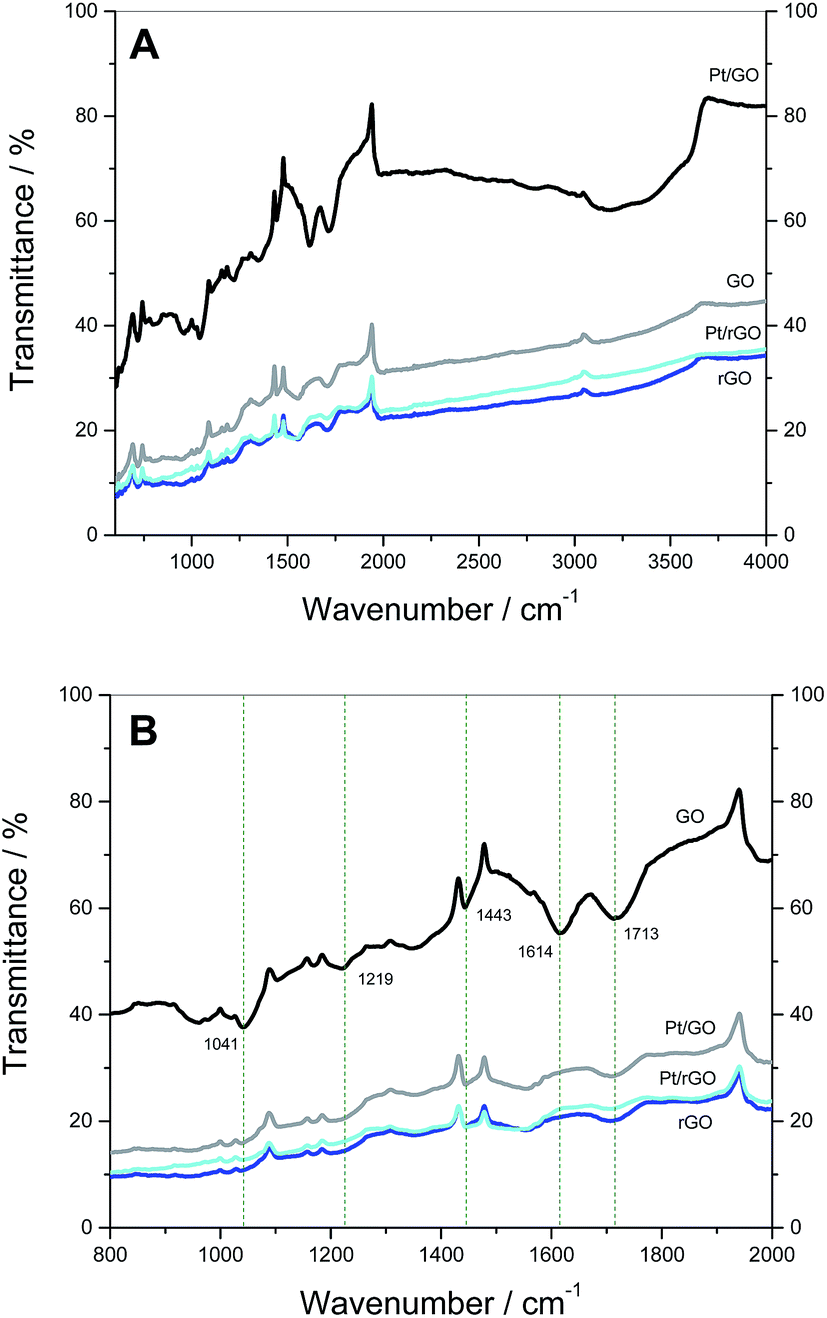 | ||
| Fig. 3 FTIR spectra of GO, rGO, Pt/GO and Pt/rGO in the range: (A) 600–4000 cm−1 and (B) 800–2000 cm−1. | ||
| Frequency, cm−1 | Bond | Functional group | Refs. |
|---|---|---|---|
| 3100–3700 | O–H stretch | Alcohol, phenol | 27, 37, 52 and 53 |
| 1713 | CO stretch |
Carbonyl | 27, 37, 52 and 53 |
| 1443 | O–H stretch | Alcohol, carboxyl | 37 |
| 1219 | C–O stretch | Epoxy | 27, 37 and 53 |
| 1041 | C–O stretch | Alkoxy | 27 and 37 |
XPS was used to quantify which groups were reduced during the procedure. According to the literature the different carbon groups contributing to the C 1s peak, namely oxygen-free C–C/CC bonds, hydroxyl C–OH (including phenols), epoxy C–O–C, carbonyl ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) CO and carboxyl HO–CO, were assigned to the corresponding binding energies 284.49, 285.86, 286.55, 287.54 and 288.94 eV, respectively.54 It has been reported that epoxy and hydroxyl groups are normally found on the basal plane of the GO layer, while carboxyls are present on the edges and ketones can be found on both basal plane and edges.55,56 Fig. 4 shows that the proportion of C–C/CC bonds in the rGO-containing samples increases with respect to GO due to the effect of L-ascorbic acid used in the GO reduction treatment. NaBH4 used in the treatment for Pt deposition also increases the proportion of C–C/CC bonds in Pt/GO with respect to GO due to collateral reduction of oxide groups, although in a lesser extent than L-ascorbic acid (see Table 3). With regard to the oxygen–carbon bonds, the epoxy groups are the main contributors to the oxide species in GO, followed by carbonyls. Nevertheless, the epoxy groups were the most susceptible to be reduced in presence of both L-ascorbic acid and sodium borohydride. It should be also mentioned that the peak at 293.3 eV present in Pt/GO and Pt/rGO is due to the presence of potassium species (K 2p3/2) remaining from the K2PtCl6 precursor used in the Pt deposition.57
CO and carboxyl HO–CO, were assigned to the corresponding binding energies 284.49, 285.86, 286.55, 287.54 and 288.94 eV, respectively.54 It has been reported that epoxy and hydroxyl groups are normally found on the basal plane of the GO layer, while carboxyls are present on the edges and ketones can be found on both basal plane and edges.55,56 Fig. 4 shows that the proportion of C–C/CC bonds in the rGO-containing samples increases with respect to GO due to the effect of L-ascorbic acid used in the GO reduction treatment. NaBH4 used in the treatment for Pt deposition also increases the proportion of C–C/CC bonds in Pt/GO with respect to GO due to collateral reduction of oxide groups, although in a lesser extent than L-ascorbic acid (see Table 3). With regard to the oxygen–carbon bonds, the epoxy groups are the main contributors to the oxide species in GO, followed by carbonyls. Nevertheless, the epoxy groups were the most susceptible to be reduced in presence of both L-ascorbic acid and sodium borohydride. It should be also mentioned that the peak at 293.3 eV present in Pt/GO and Pt/rGO is due to the presence of potassium species (K 2p3/2) remaining from the K2PtCl6 precursor used in the Pt deposition.57
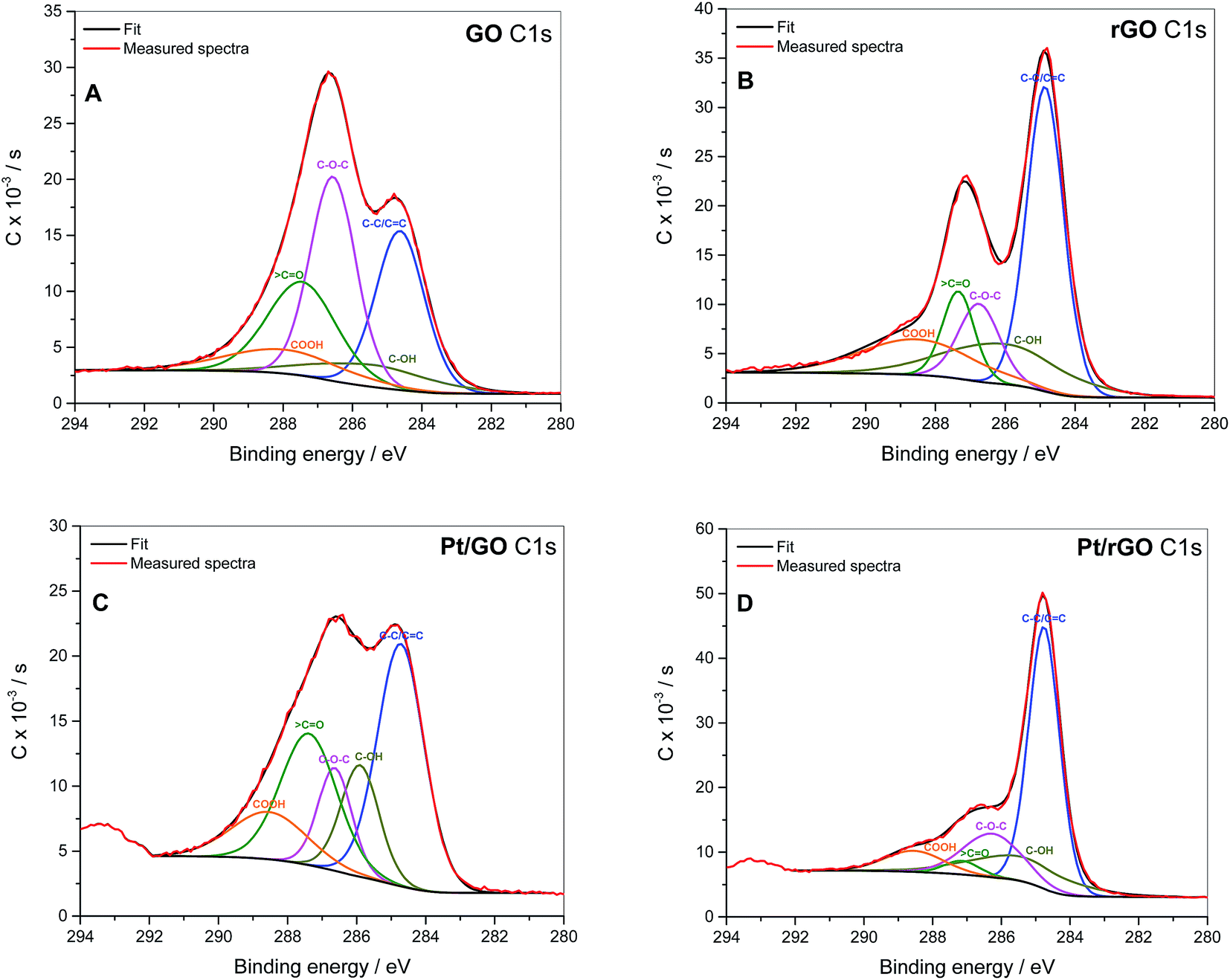 | ||
| Fig. 4 C 1s core-level XPS spectra of: (A) GO, (B) rGO, (C) Pt/GO and (D) Pt/rGO. | ||
| GO | Pt/GO | rGO | Pt/rGO | |
|---|---|---|---|---|
| C–C/CC |
26.7 | 37.8 | 43.2 | 54.8 |
| C–OH | 8.9 | 14.2 | 18.4 | 16.5 |
| C–O–C | 32.5 | 11.5 | 12.2 | 17.2 |
| CO |
22.3 | 24.7 | 10.7 | 3.2 |
| COOH | 9.7 | 11.9 | 15.5 | 8.3 |
3.2. Electrochemical characterisation
In this section the behaviour of the catalyst-supports towards the ORR in alkaline media is studied. The ORR takes place in alkaline media according to the overall, 4-electron reaction (1):58| O2 + 2H2O + 4e− → 4OH− E0 = +0.401 V vs. SHE | (1) |
However, the reaction can also proceed stepwise, via the following 2-electron processes:59
| O2 + H2O + 2e− → HO2− + OH− E0 = −0.076 V vs. SHE | (2) |
| HO2− + H2O + 2e− → 3OH− E0 = +0.878 V vs. SHE | (3) |
| 2HO2− → 2OH− + O2 | (4) |
As it can be seen, the alternative 2-step pathway produces peroxide intermediates. This is a problem in alkaline media, as the lower working potential of the ORR electrode in this environment favours the desorption of HO2−, whereas in acidic conditions the high working potential avoids the peroxide desorption thus facilitating the further reduction to OH−.60 The ORR takes place mainly through HO2− formation at graphite and most carbon surfaces, whereas it follows the 4-electron pathway in Pt catalysts.58 As a result, in catalytic systems formed by Pt particles deposited on carbon supports it is expected to find a mixed behaviour between the two proposed mechanisms, depending on the behaviour of the carbon support. In order to determine which mechanism is predominant the peroxide intermediate production should be monitored. This can be done with a RRDE, setting the ring potential at the peroxide diffusion-limiting value. This methodology has been successfully proven in former work carried out by Damjanovic and co-workers61 and then developed by subsequent investigations.62–64
In order to compare the ORR activity of each catalyst, voltammograms were measured at a voltage scan rate of 25 mV s−1 over the range from +0.4 to −1.0 V (vs. Ag/AgCl 3 M ref. electrode) in oxygen-saturated 0.1 M KOH solution using a RRDE modified with the catalyst, as described in Section 2.2. Fig. 5A shows the voltammograms of the different catalysts at a rotation speed of 1600 rpm. If we consider the ORR activity in terms of more positive onset potential it can be seen that this parameter is in good correlation with the values of ECSA calculated (Table 4). It is observed that the ECSA of Pt/GO and Pt/rGO is very low, which is attributed to the presence of oxide groups affecting negatively to the catalytic activity of the Pt particles, in spite of the good particle size distribution displayed in Fig. 2. A similar pattern is shown in terms of half-wave potential, except for the case of Pt/MWCNT and Pt/rGO which swap their positions with respect to the onset values. The behaviour of Pt/MWCNT at more negative potentials could be influenced by the presence of metal impurities in its structure, as we will analyse later.
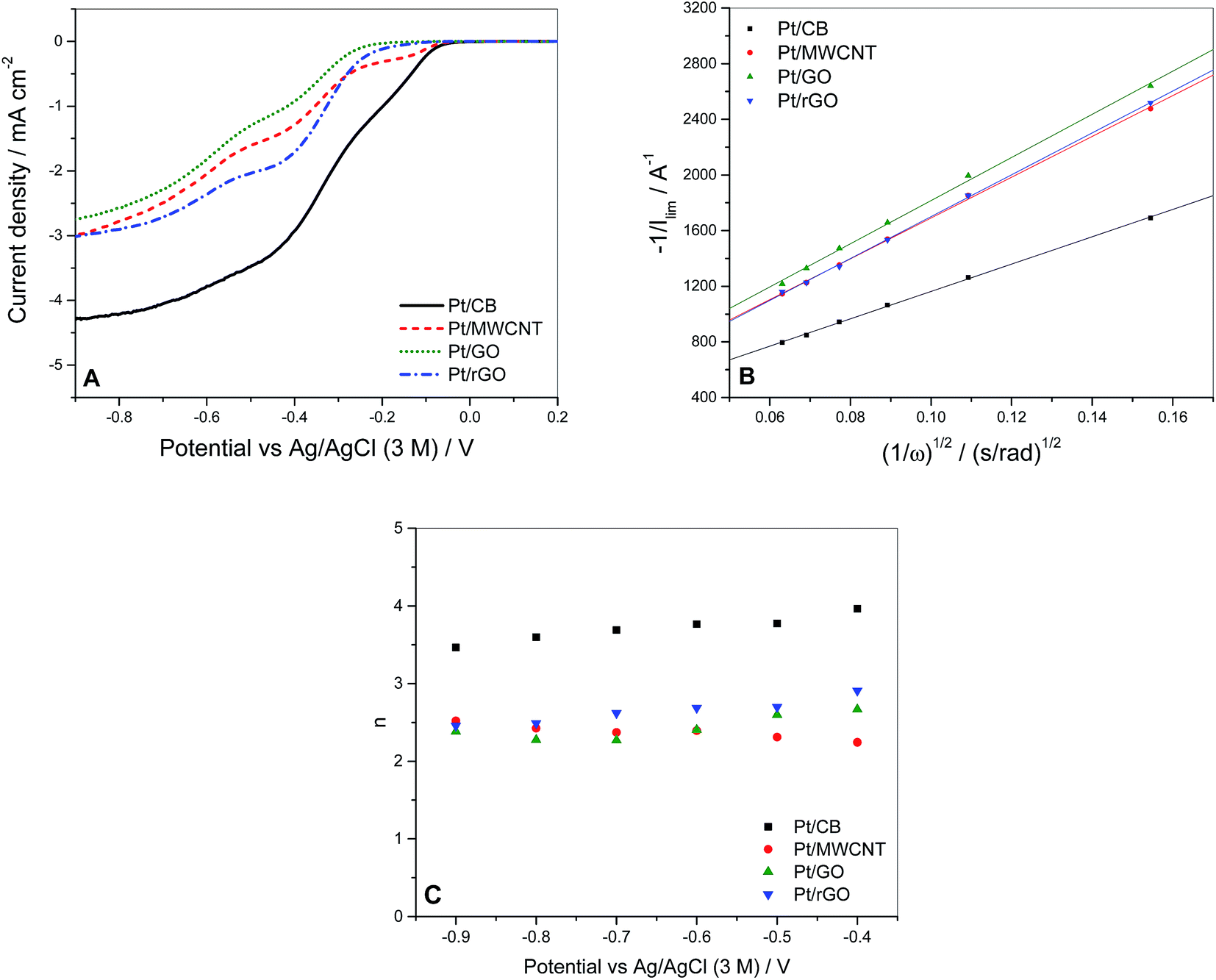 | ||
| Fig. 5 (A) LSV measurements of the 4 different synthesised catalysts at a rotation speed of 1600 rpm. (B) Koutecky–Levich plots of the 4 catalysts from the values of measured current at −0.9 V. (C) Values of n obtained from the application of the Koutecky–Levich equation to the LSV data versus the potential. | ||
| Catalyst | Onset potential/V | Half-wave potential/V | ECSA/m2 g−1 |
|---|---|---|---|
| Pt/CB | −0.05 | −0.33 | 20.6 |
| Pt/MWCNT | −0.07 | −0.46 | 15.3 |
| Pt/GO | −0.21 | −0.52 | 1.8 |
| Pt/rGO | −0.13 | −0.38 | 3.5 |
To investigate the dominant ORR mechanism the number of electrons n involved in the electrochemical reaction was determined via Koutecky–Levich (KL) analysis (eqn (5)):65
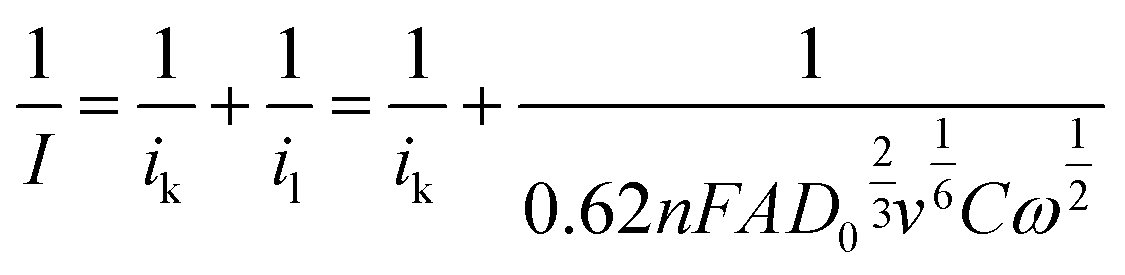 | (5) |
To gain more information on the mechanism, a RRDE was used to directly measure the rate of peroxide formation. Here the ORR takes place at the GC disk (where the different catalysts were deposited) and the concentric Pt ring detects the peroxide produced. In Fig. 6 it is shown the RRDE plots for each catalyst, showing the measured disk current ID at the bottom and the measured ring current IR at the top.
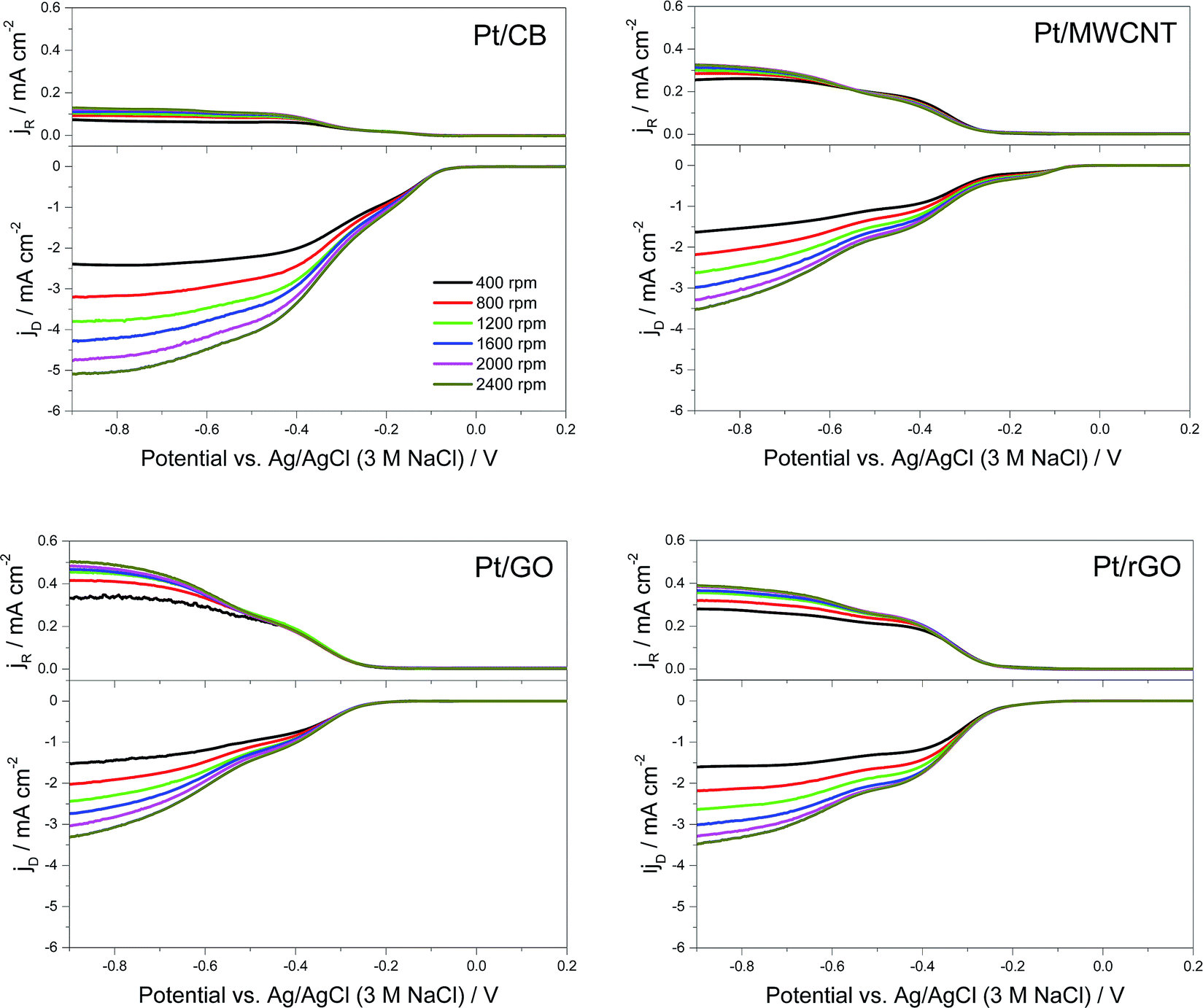 | ||
| Fig. 6 Background-subtracted RRDE measurements at different rotation rates for the 4 catalysts at loadings of 20 μg Pt/C cm−2 on 0.2475 cm2 area GC working electrode in O2-saturated 0.1 M KOH at 25 °C. IR ring current (ring potential fixed at +0.47 V) is displayed at the top (ring area 0.1866 cm2) and ID disk current is displayed at the bottom. | ||
From these graphs it can be calculated the number of electrons transferred in the electrochemical reaction (n) from the values of IR and ID using the eqn (6):64
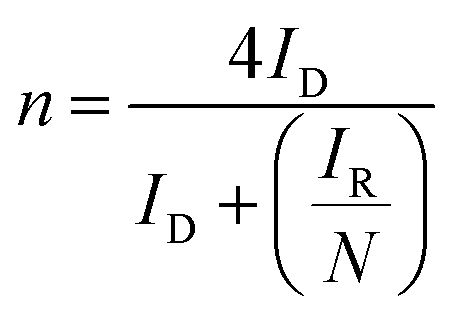 | (6) |
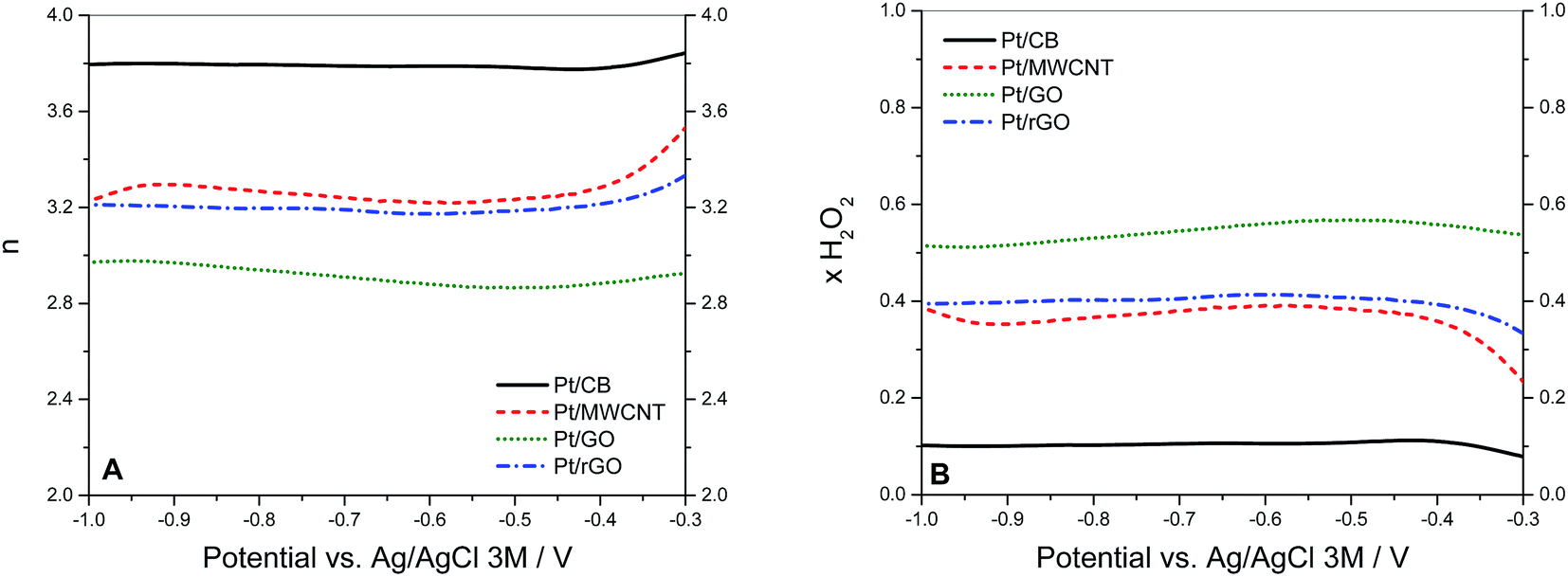 | ||
| Fig. 7 (A) Values of n vs. the potential calculated from the RRDE measurements at 1600 rpm using eqn (6). (B) Fraction of H2O2 formed during O2 reduction the 4 catalysts at 25 °C in 0.1 KOH saturated with O2. Calculated from RRDE measurements at 1600 rpm using eqn (7) with N = 0.37. | ||
If these values are compared to those obtained by the KL equation it can be seen that the RRDE method provides higher values of n. For Pt/CB the ORR involved 3.8 electrons which is independent of the potential applied in the range between −0.4 and −1.0 V. This stability of n in that potential range was also shown by Pt/rGO with a constant value of 3.2. The value of n varied in the cases of Pt/GO and Pt/MWCNT, indicating that the ORR was more sensitive to changes in the potential, especially for the later. These values of n are closely related to the selectivity of the reaction which can be quantified by the fraction of peroxide intermediate produced in the reaction (XH2O2) using the eqn (7):38
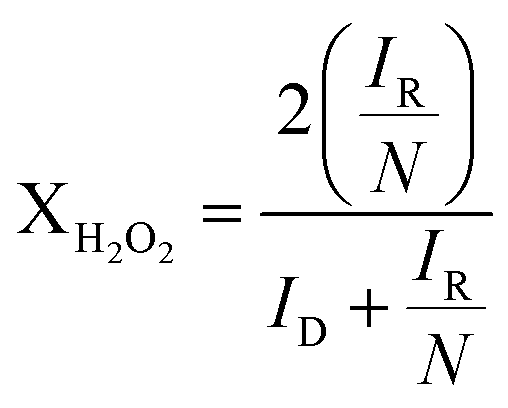 | (7) |
The values of XH2O2 versus the potential applied at 1600 rpm are plotted in Fig. 7B. The peroxide intermediate production is around 10% for Pt/CB, 35% for Pt/MWCNT, 40% for Pt/rGO and 55% for Pt/GO, which is in accordance with the catalytic behaviour showed by each catalyst in previous measurements.
The variation of the values of n and XH2O2 with the applied voltage is significant in the case of Pt/MWCNT. There could be two different explanations for this. First, this could be attributed to metal impurities remaining in the MWCNTs from the fabrication process.67,68 In the present investigation, XRF analysis has been carried out in order to determine the nature of these metal impurities,69 showing that Fe is by far the main contributor with 11130 ± 90 ppm, followed in a lesser extent by Cd (270 ± 10 ppm), Ni (200 ± 20 ppm), In (130 ± 10 ppm), Zn (130 ± 10 ppm) and Pb (110 ± 30 ppm). All these impurities are also found in the rest of the carbon supports but only Fe impurities are 10 times higher in MWCNT with respect to the others, suggesting that Fe plays an important role on the catalytic activity of MWCNT.70 The fact of that no Fe impurities are detected in XPS studies (which is a surface technique) carried out on the Pt/MWCNT catalyst clearly indicates that the Fe particles are embedded inside the nanotube structure.71 Most of available literature on the topic points to that these Fe impurities are in the form of Fe3O4,72 being this specie more catalytically active at potentials around −0.8 V (vs. Ag/AgCl).73 This could explain why the value of n is higher (and consequently XH2O2 is lower) in the case of Pt/MWCNT at more negative potentials. The second possibility is related to the role of the 1D structure of MWCNT on the catalytic performance. It has been proposed that in some types of carbon nanotubes the number of electrons transferred during the ORR is function of the potential applied, increasing the value of n at more negative potentials.74 Nevertheless, Pumera and Wang demonstrated that the catalytic activity of these supposedly “metal-free” materials is in fact due to metal impurities remaining from the process of fabrication.73
We can also observe that Pt/GO undergoes a considerably lower catalytic activity in spite of its relatively higher Pt loading (around 19%) compared to the rest of the catalysts (around 15%). The reason of this poor performance of the Pt/GO catalyst should be obviously related to the intrinsic characteristics of the carbon support. Especially remarkable are the differences of catalytic activity in Pt/GO and Pt/rGO taking into account that they were synthesised from the same GO carbon support, but subjected to a previous reductive method in the case of rGO. XPS studies (see Fig. 4C and D) demonstrated that the oxide groups had been removed in a larger extent in Pt/rGO with respect to Pt/GO. Although it is commonly accepted that the electrical conductivity of GO can be improved by partially restoring the sp2 structure through oxide group elimination (especially epoxy and hydroxyl species located on the basal plane),28 this is not our case. AC impedance measurements carried out as described in Experimental section 2.2, show that the ohmic resistance of the Pt/GO layer deposited on the RDE is 2.8 ± 0.7 Ω versus 3.2 ± 0.4 Ω for Pt/rGO, indicating that the oxide groups in the basal plane (epoxy and hydroxyl) implied in the electronic conductivity were less affected by the reduction treatment. This is supported by the observation (Table 3) of that the main oxide groups being removed in Pt/rGO with respect to Pt/GO were carbonyl and carboxyl, which are located on the edges. Another study demonstrates the influence of quinones (containing the carbonyl group) on the ORR performance,75 confirming that the oxygen reduction in rGO takes place mainly on the active reduced sites located on the edges of the graphene layers.
In addition, the values of ohmic resistance obtained for Pt/CB and Pt/MWCNT were 8.1 ± 1.2 Ω and 6.5 ± 1.4 Ω respectively, which confirms that the conductivity played just a secondary role on the catalytic activity observed. Considering the number of electrons involved in the ORR, the higher value of n calculated for Pt/CB suggests that the catalytic activity of the Pt dominates the ORR in Pt/CB, since it is well-known that the oxygen reduction follows a 4-electron mechanism on Pt surfaces in both alkaline and acid media, while the 2-electron mechanism is preferred on carbon surfaces.58,76 For example, Mao et al. reported that the ORR takes place in MWCNT by the 2-electron mechanism in alkaline media,19 while Papakonstantinou et al. concluded that electrochemically reduced graphene oxide follows a more predominant 2e− pathway.75 Likewise, the lower value of n for Pt on MWCNT, GO and rGO reveals a greater contribution of the carbon-mediated pathway and thus reflects that these forms of carbon are more active towards the ORR than CB. In conclusion, in the supports where the carbon configuration is derived from the graphene structure (MWCNT, GO and rGO), the ORR seems to take place on both carbon and Pt particles, thus showing a mixed behaviour between the two proposed mechanisms.
4. Conclusions
An integral study on the behaviour towards the oxygen reduction of catalysts with different carbon supports has been carried out in alkaline media. TGA analysis demonstrated that the reduction of potassium hexachloroplatinate with sodium borohydride is an efficient method for Pt deposition, whereas XPS analysis proved that reduction of GO with L-ascorbic acid is an effective method to eliminate the characteristic oxide species present in GO, especially epoxy groups. It is also concluded that the Pt/CB catalyst performs a quasi 4-electron pathway for the oxygen reduction, whereas Pt/MWCNT, Pt/GO and Pt/rGO underwent a mixed behaviour between the 1-step and the 2-step proposed mechanisms. Especially Pt/GO yielded a poorer performance compared to Pt/rGO, presumably due to the presence of carbonyl and carboxyl species on the edges hindering the oxygen reduction reaction. These results were obtained by indirect calculation using the Koutecky–Levich equation and confirmed by direct determination of peroxide intermediate formation with RRDE measurements.Acknowledgements
The authors would like to thank the EPSRC for its financial support and the members of the Hydrogen and Fuel Cell Research Centre at University of Birmingham for their valuable help during the experimental work. Furthermore, we would like also to express our gratitude to the National EPSRC XPS Users' Service (NEXUS) at Newcastle University, an EPSRC Mid-Range Facility, for their collaboration with the XPS measurements, as well as the Nanoscale and Microscale Research Centre (NMRC) at The University of Nottingham, especially to Dr Michael Fay, for their support with the TEM imaging.References
- E. Bailón-García, F. Maldonado-Hódar, A. Pérez-Cadenas and F. Carrasco-Marín, Catalysts Supported on Carbon Materials for the Selective Hydrogenation of Citral, Catalysts, 2013, 3(4), 853–877 CrossRef.
- S. Sharma and B. G. Pollet, Support materials for PEMFC and DMFC electrocatalysts – a review, J. Power Sources, 2012, 208, 96–119 CrossRef CAS.
- M. Watanabe, H. Sei and P. Stonehart, The influence of platinum crystallite size on the electroreduction of oxygen, J. Electroanal. Chem., 1989, 261(2), 375–387 CrossRef CAS.
- M. a. Fraga, E. Jordão, M. J. Mendes, M. M. a. Freitas, J. L. Faria and J. L. Figueiredo, Properties of Carbon-Supported Platinum Catalysts: Role of Carbon Surface Sites, J. Catal., 2002, 209(2), 355–364 CrossRef CAS.
- Q. Wu, Z. Rao, L. Yuan, L. Jiang, G. Sun and J. Ruan, et al., Carbon supported PdO with improved activity and stability for oxygen reduction reaction in alkaline solution, Electrochim. Acta, 2014, 150, 157–166 CrossRef CAS.
- L. Jiang, A. Hsu, D. Chu and R. Chen, A highly active Pd coated Ag electrocatalyst for oxygen reduction reactions in alkaline media, Electrochim. Acta, 2010, 55(15), 4506–4511 CrossRef CAS.
- F. H. B. Lima, J. Zhang, M. H. Shao, K. Sasaki, M. B. Vukmirovic and E. a. Ticianelli, et al., Catalytic activity – d-band center correlation for the O2 reduction reaction on platinum in alkaline solutions, J. Phys. Chem. C, 2007, 111(1), 404–410 CAS.
- X. Li, B. N. Popov, T. Kawahara and H. Yanagi, Non-precious metal catalysts synthesized from precursors of carbon, nitrogen, and transition metal for oxygen reduction in alkaline fuel cells, J. Power Sources, 2011, 196(4), 1717–1722 CrossRef CAS.
- S. D. Poynton, J. P. Kizewski, R. C. T. Slade and J. R. Varcoe, Novel electrolyte membranes and non-Pt catalysts for low temperature fuel cells, Solid State Ionics, 2010, 181(3–4), 219–222 CrossRef CAS.
- H. Bunazawa and Y. Yamazaki, Influence of anion ionomer content and silver cathode catalyst on the performance of alkaline membrane electrode assemblies (MEAs) for direct methanol fuel cells (DMFCs), J. Power Sources, 2008, 182(1), 48–51 CrossRef CAS.
- C. Coutanceau, L. Demarconnay, C. Lamy and J. M. Léger, Development of electrocatalysts for solid alkaline fuel cell (SAFC), J. Power Sources, 2006, 156(1), 14–19 CrossRef CAS.
- L. Li, X. Yuan, Z. Ma and Z.-F. Ma, Properties of Pyrolyzed Carbon-Supported Cobalt-Polypyrrole as Electrocatalyst toward Oxygen Reduction Reaction in Alkaline Media, J. Electrochem. Soc., 2015, 162(4), F359–F365 CrossRef CAS.
- Y. Yuan, J. Ahmed and S. Kim, Polyaniline/carbon black composite-supported iron phthalocyanine as an oxygen reduction catalyst for microbial fuel cells, J. Power Sources, 2011, 196(3), 1103–1106 CrossRef CAS.
- W. Sun, A. Hsu and R. Chen, Carbon-supported tetragonal MnOOH catalysts for oxygen reduction reaction in alkaline media, J. Power Sources, 2011, 196(2), 627–635 CrossRef CAS.
- E. Antolini, F. Cardellini, E. Giacometti and G. Squadrito, Study on the formation of Pt/C catalysts by non-oxidized active carbon support and a sulfur-based reducing agent, J. Mater. Sci., 2002, 37(1), 133–139 CrossRef CAS.
- C. K. Poh, S. H. Lim, H. Pan, J. Lin and J. Y. Lee, Citric acid functionalized carbon materials for fuel cell applications, J. Power Sources, 2008, 176(1), 70–75 CrossRef CAS.
- Y. Takasu, T. Kawaguchi, W. Sugimoto and Y. Murakami, Effects of the surface area of carbon support on the characteristics of highly-dispersed Pt–Ru particles as catalysts for methanol oxidation, Electrochim. Acta, 2003, 48(25–26), 3861–3868 CrossRef CAS.
- V. Rao, P. a. Simonov, E. R. Savinova, G. V. Plaksin, S. V. Cherepanova and G. N. Kryukova, et al., The influence of carbon support porosity on the activity of PtRu/Sibunit anode catalysts for methanol oxidation, J. Power Sources, 2005, 145(2), 178–187 CrossRef CAS.
- M. Zhang, Y. Yan, K. Gong, L. Mao, Z. Guo and Y. Chen, Electrostatic layer-by-layer assembled carbon nanotube multilayer film and its electrocatalytic activity for O2 reduction, Langmuir, 2004, 20(20), 8781–8785 CrossRef CAS PubMed.
- L. Tammeveski, H. Erikson, A. Sarapuu, J. Kozlova, P. Ritslaid and V. Sammelselg, et al., Electrocatalytic oxygen reduction on silver nanoparticle/multi-walled carbon nanotube modified glassy carbon electrodes in alkaline solution, Electrochem. Commun., 2012, 20(1), 15–18 CrossRef CAS.
- X. Yu and A. Manthiram, MnNiCoO/n 4/n/N-MWCNT nanocomposite catalyst with high selectivity in membraneless direct formate fuel cells and bifunctional activity for oxygen electrochemistry, Catal. Sci. Technol., 2015, 5, 2072–2075 CAS.
- W. Chen, Y. Zhang and X. Wei, Catalytic performances of PdNi/MWCNT for electrooxidations of methanol and ethanol in alkaline media, Int. J. Hydrogen Energy, 2015, 40(2), 1154–1162 CrossRef CAS.
- A. Dumitru, M. Mamlouk and K. Scott, Effect of different chemical modification of carbon nanotubes for the oxygen reduction reaction in alkaline media, Electrochim. Acta, 2014, 135, 428–438 CrossRef CAS.
- A. Marinkas, F. Arena, J. Mitzel, G. M. Prinz, A. Heinzel and V. Peinecke, et al., Graphene as catalyst support: the influences of carbon additives and catalyst preparation methods on the performance of PEM fuel cells, Carbon, 2013, 58(i), 139–150 CrossRef CAS.
- M. H. Seo, S. M. Choi, J. K. Seo, S. H. Noh, W. B. Kim and B. Han, The graphene-supported palladium and palladium-yttrium nanoparticles for the oxygen reduction and ethanol oxidation reactions: experimental measurement and computational validation, Appl. Catal., B, 2013, 129, 163–171 CrossRef CAS.
- C. Mattevi, G. Eda, S. Agnoli, S. Miller, K. A. Mkhoyan and O. Celik, et al., Evolution of electrical, chemical, and structural properties of transparent and conducting chemically derived graphene thin films, Adv. Funct. Mater., 2009, 19(16), 2577–2583 CrossRef CAS.
- J. Song, X. Wang and C.-T. Chang, Preparation and characterization of graphene oxide, J. Nanomater., 2014, 2014, 1–6 Search PubMed.
- S. Pei and H. M. Cheng, The reduction of graphene oxide, Carbon, 2012, 50(9), 3210–3228 CrossRef CAS.
- I. Jung, D. a. Dikin, R. D. Piner and R. S. Ruoff, Tuneable Electrical Conductivity of Individual Graphene Oxide Sheets Reduced at “Low” Temperatures, Nano Lett., 2008, 8(12), 4283–4287 CrossRef CAS PubMed.
- W. J. Basirun, M. Sookhakian, S. Baradaran, Z. Endut, M. R. Mahmoudian and M. Ebadi, et al., Graphene oxide electrocatalyst on MnO2 air cathode as an efficient electron pump for enhanced oxygen reduction in alkaline solution, Sci. Rep., 2015, 5, 9108 CrossRef CAS PubMed.
- A. Y. S. Eng and M. Pumera, Direct voltammetry of colloidal graphene oxides, Electrochem. Commun., 2014, 43, 87–90 CrossRef CAS.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, The chemistry of graphene oxide, Chem. Soc. Rev., 2010, 39(1), 228–240 RSC.
- E. J. Lim, S. M. Choi, M. H. Seo, Y. Kim, S. Lee and W. B. Kim, Highly dispersed Ag nanoparticles on nanosheets of reduced graphene oxide for oxygen reduction reaction in alkaline media, Electrochem. Commun., 2013, 28, 100–103 CrossRef CAS.
- A. Ambrosi, A. Bonanni, Z. Sofer, J. S. Cross and M. Pumera, Electrochemistry at chemically modified graphenes, Chem.–Eur. J., 2011, 17(38), 10763–10770 CrossRef CAS PubMed.
- M. J. Fernández-Merino, L. Guardia, J. I. Paredes, S. Villar-Rodil, P. Solís-Fernández and a. Martínez-Alonso, et al., Vitamin C is an ideal substitute for hydrazine in the reduction of graphene oxide suspensions, J. Phys. Chem. C, 2010, 114(14), 6426–6432 Search PubMed.
- B. Fang, N. K. Chaudhari, M.-S. Kim, J. H. Kim and J.-S. Yu, Homogeneous deposition of platinum nanoparticles on carbon black for proton exchange membrane fuel cell, J. Am. Chem. Soc., 2009, 131(42), 15330–15338 CrossRef CAS PubMed.
- J. Zhang, H. Yang, G. Shen, P. Cheng, J. Zhang and S. Guo, Reduction of graphene oxide via L-ascorbic acid, Chem. Commun., 2010, 46(7), 1112–1114 RSC.
- J. E. Newton, J. a. Preece, N. V. Rees and S. L. Horswell, Nanoparticle catalysts for proton exchange membrane fuel cells: can surfactant effects be beneficial for electrocatalysis?, Phys. Chem. Chem. Phys., 2014, 16(23), 11435–11446 RSC.
- Y. Garsany, I. L. Singer and K. E. Swider-Lyons, Impact of film drying procedures on RDE characterization of Pt/VC electrocatalysts, J. Electroanal. Chem., 2011, 662(2), 396–406 CrossRef CAS.
- J. Sunarso, A. a. J. Torriero, W. Zhou, P. C. Howlett and M. Forsyth, Oxygen reduction reaction activity of La-based perovskite oxides in alkaline medium: a thin-film rotating ring-disk electrode study, J. Phys. Chem. C, 2012, 116(9), 5827–5834 CAS.
- E. F. Antunes, V. G. De Resende, U. a. Mengui, J. B. M. Cunha, E. J. Corat and M. Massi, Analyses of residual iron in carbon nanotubes produced by camphor/ferrocene pyrolysis and purified by high temperature annealing, Appl. Surf. Sci., 2011, 257(18), 8038–8043 CrossRef CAS.
- Y. Qiu, F. Guo, R. Hurt and I. Külaots, Explosive thermal reduction of graphene oxide-based materials: mechanism and safety implications, Carbon, 2014, 72, 215–223 CrossRef CAS PubMed.
- R. Yu, L. Chen, Q. Liu, J. Lin, K. Tan and S. C. Ng, et al., Platinum Deposition on Carbon Nanotubes via Chemical Modification, Chem. Mater., 1998, 10, 718–722 CrossRef CAS.
- F. Li, Y. Guo, T. Wu, Y. Liu, W. Wang and J. Gao, Platinum nano-catalysts deposited on reduced graphene oxides for alcohol oxidation, Electrochim. Acta, 2013, 111, 614–620 CrossRef CAS.
- R. Nie, J. Wang, L. Wang, Y. Qin, P. Chen and Z. Hou, Platinum supported on reduced graphene oxide as a catalyst for hydrogenation of nitroarenes, Carbon, 2012, 50(2), 586–596 CrossRef CAS.
- M. Kheirmand and A. Eshghi, Electro deposition of platinum nanoparticles on reduced graphene oxide as an efficient catalyst for oxygen reduction reaction, Iran. J. Hydrogen Fuel Cell, 2015, 1, 7–12 Search PubMed.
- J. W. Ma, a. L. Y. Habrioux, G. Ramos-Sanchez, L. Calvillo and G. Granozzi, et al., Electronic interaction between platinum nanoparticles and nitrogen-doped reduced graphene oxide: effect on the oxygen reduction reaction, J. Mater. Chem. A, 2015, 3(22), 11891–11904 CAS.
- M. Nesselberger, S. Ashton, J. C. Meier, I. Katsounaros, K. J. J. Mayrhofer and M. Arenz, The particle size effect on the oxygen reduction reaction activity of Pt catalysts: influence of electrolyte and relation to single crystal models, J. Am. Chem. Soc., 2011, 133(43), 17428–17433 CrossRef CAS PubMed.
- K. Kinoshita, Particle Size Effects for Oxygen Reduction on Highly Dispersed Platinum in Acid Electrolytes, J. Electrochem. Soc., 1990, 137(3), 845 CrossRef CAS.
- H. Yano, J. Inukai, H. Uchida, M. Watanabe, P. K. Babu and T. Kobayashi, et al., Particle-size effect of nanoscale platinum catalysts in oxygen reduction reaction: an electrochemical and 195Pt EC-NMR study, Phys. Chem. Chem. Phys., 2006, 8(42), 4932–4939 RSC.
- E. Fuente, J. a. Menéndez, M. a. Díez, D. Suárez and M. a. Montes-Morán, Infrared Spectroscopy of Carbon Materials: A Quantum Chemical Study of Model Compounds, J. Phys. Chem. B, 2003, 107, 6350–6359 CrossRef CAS.
- T. Szabó, O. Berkesi and I. Dékány, DRIFT study of deuterium-exchanged graphite oxide, Carbon, 2005, 43(15), 3186–3189 CrossRef.
- V. Loryuenyong, K. Totepvimarn, P. Eimburanapravat, W. Boonchompoo and A. Buasri, Preparation and Characterization of Reduced Graphene Oxide Sheets via Water-Based Exfoliation and Reduction Methods, Adv. Mater. Sci. Eng., 2013, 2013, 923403 Search PubMed.
- A. Ganguly, S. Sharma, P. Papakonstantinou and J. Hamilton, Probing the Thermal Deoxygenation of Graphene Oxide using High Resolution In Situ X-Ray based Spectroscopies, J. Phys. Chem., 2011, 17009–17019 CAS.
- A. Bagri, C. Mattevi, M. Acik, Y. J. Chabal, M. Chhowalla and V. B. Shenoy, Structural evolution during the reduction of chemically derived graphene oxide, Nat. Chem., 2010, 2(7), 581–587 CrossRef CAS PubMed.
- T. Szab, O. Berkesi, P. Forg, K. Josepovits, Y. Sanakis and D. Petridis, et al., Evolution of Surface Functional Groups in a Series of Progressively Oxidized Graphite Oxides, Chem. Mater., 2006, 18, 2740–2749 CrossRef.
- S. Li, E. T. Kang, K. G. Neoh, Z. H. Ma, K. L. Tan and W. Huang, In situ XPS studies of thermally deposited potassium on poly(p-phenylene vinylene) and its ring-substituted derivatives, Appl. Surf. Sci., 2001, 181(3–4), 201–210 CrossRef CAS.
- E. Yeager, Dioxygen electrocatalysis: mechanisms in relation to catalyst structure, J. Mol. Catal., 1986, 38(1–2), 5–25 CrossRef CAS.
- X. Ge, A. Sumboja, D. Wuu, T. An, B. Li and F. W. T. Goh, et al., Oxygen Reduction in Alkaline Media: From Mechanisms to Recent Advances of Catalysts, ACS Catal., 2015, 5(8), 4643–4667 CrossRef CAS.
- J. S. Spendelow and A. Wieckowski, Electrocatalysis of oxygen reduction and small alcohol oxidation in alkaline media, Phys. Chem. Chem. Phys., 2007, 9(21), 2654–2675 RSC.
- A. Damjanovic, M. A. Genshaw and J. O. Bockris, The mechanism of oxygen reduction at platinum in alkaline solutions with special reference to H2O2, J. Electrochem. Soc., 1967, 114(11), 1107–1112 CrossRef CAS.
- K. F. Blurton and E. McMullin, The Cathodic Reduction of Oxygen on Platinum in Alkaline Solutions, J. Electrochem. Soc., 1969, 116(11), 1476 CrossRef CAS.
- N. M. Markovic, H. a. Gasteiger and P. N. Ross, Oxygen Reduction on Platinum Low-Index Single-Crystal Surfaces in Sulfuric Acid Solution: Rotating Ring-Pt(hkZ) Disk Studies, J. Phys. Chem., 1995, 99(11), 3411 CrossRef CAS.
- E. Claude, T. Addou, J. Latour and P. Aldebert, A new method for electrochemical screening based on the rotating ring disc electrode and its application to oxygen reduction catalysts, J. Appl. Electrochem., 1998, 28(1), 57–64 CrossRef CAS.
- A. J. Bard, L. R. Faulkner, N. York, C. Bullet, W. Brisbane and S. E. Toronto, Electrochemical Methods Fundamentals and Applications, Electrochemistry, ed. I. Faulkner and R. Larry, 1944, pp. 3–7 Search PubMed.
- N. Wakabayashi, M. Takeichi, M. Itagaki, H. Uchida and M. Watanabe, Temperature-dependence of oxygen reduction activity at a platinum electrode in an acidic electrolyte solution investigated with a channel flow double electrode, J. Electroanal. Chem., 2005, 574(2), 339–346 CrossRef CAS.
- Y. Li, W. Zhou, H. Wang, L. Xie, Y. Liang and F. Wei, et al., An oxygen reduction electrocatalyst based on carbon nanotube-graphene complexes, Nat. Nanotechnol., 2012, 7, 394–400 CrossRef CAS PubMed.
- P. Gayathri and A. Senthil, An Iron Impurity in Multiwalled Carbon Nanotube Complexes with Chitosan that Biomimics the Heme-Peroxidase Function, Chem.–Eur. J., 2013, 19, 17103–17112 CrossRef CAS PubMed.
- V. A. Online, M. Pumera, E. Lay, K. Chng and H. L. Poh, Purification of carbon nanotubes by high temperature chlorine gas treatment, Phys. Chem. Chem. Phys., 2013, 15, 5615–5619 RSC.
- K. M. Samant, V. S. Joshi and K. R. Patil, Role of iron oxide impurities in electrocatalysis by multiwall carbon nanotubes: an investigation using a novel magnetically modified ITO, Bull. Mater. Sci., 2014, 37(2), 221–226 CrossRef CAS.
- T. Kolodiazhnyi and M. Pumera, Towards an Ultrasensitive Method for the Determination of Metal Impurities in Carbon Nanotubes, Small, 2008, 4(9), 1476–1484 CrossRef CAS PubMed.
- J. Kruusma, N. Mould, K. Jurkschat, A. Crossley and C. E. Banks, Single walled carbon nanotubes contain residual iron oxide impurities which can dominate their electrochemical activity, Electrochem. Commun., 2007, 9, 2330–2333 CrossRef CAS.
- L. Wang and M. Pumera, Residual metallic impurities within carbon nanotubes play a dominant role in supposedly “metal-free” oxygen reduction reactions, Chem. Commun., 2014, 50, 12662–12664 RSC.
- G. Ju and K. Tammeveski, Electroreduction of oxygen on multi-walled carbon nanotubes modified highly oriented pyrolytic graphite electrodes in alkaline solution, J. Electroanal. Chem., 2006, 597, 119–126 CrossRef.
- S. K. Bikkarolla, P. Cumpson, P. Joseph and P. Papakonstantinou, Oxygen reduction reaction by electrochemically reduced graphene oxide, Faraday Discuss., 2014, 173, 415–428 RSC.
- C. Song and J. Zhang, Electrocatalytic Oxygen Reduction Reaction, PEM Feul Cell Electrocatal Catal Layers Fundam Appl, 2008, pp. 89–134 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |