
Benzothiophene-flanked diketopyrrolopyrrole polymers: impact of isomeric frameworks on carrier mobilities†
Abstract
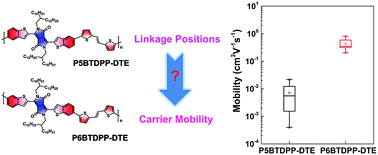
Exploring new building blocks for solution-processable polymeric semiconductors has attracted much attention. We herein develop two isomeric benzothiophene-flanked diketopyrrolopyrrole polymers with different linkage positions and further systematically study the electronic structures, optical properties, and field-effect characteristics. Both polymers exhibit typical p-type transport characteristics with the highest mobility being up to 0.80 cm2 V−1 s−1. Our results suggest that the isomeric backbones for conjugated polymers greatly affect charge transport properties.
 Please wait while we load your content...
Please wait while we load your content...

