
Sulfonamide-1,3,5-triazine–thiazoles: discovery of a novel class of antidiabetic agents via inhibition of DPP-4
Hai-De Gao*,
Peng Liu,
Yang Yang and
Fang Gao
Department of Hepatopancreatobiliary and Splenic Medicine, Affiliated Hospital, Logistics University of Chinese People's Armed Police Force, Tianjin 300162, China. E-mail: gaohaide2012@hotmail.com; Fax: +86-22-60578765; Tel: +86-22-60578765
First published on 9th August 2016
Abstract
Dipeptidyl peptidase-4 (DPP-4) inhibitors are a novel class of antidiabetic drugs used for treating type 2 diabetes mellitus. In the present study, a novel hybrid sulphonamide-1,3,5-triazine–thiazole derivatives were synthesized and characterized by 1H-NMR, 13C-NMR, FT-IR, mass spectroscopy and elemental analysis. The result showed that among the tested compounds, 8c was found to be a more potent inhibitor of DPP-4 (2.32 nM) than the alogliptin standard. Moreover, molecular docking results showed that ligand 8c was efficiently docked into the active site of the catalytic triad of Ser630, Asp708 and His740, encompassing both the S1 and S2 pocket with a CDOCKER interaction energy of 57.80. The in vitro results were further substantiated by in vivo blood glucose lowering effects in experimental subjects. The results of the investigation showed that compound 8c exhibited concentration-dependent improvement of glucose tolerance in ICR after oral administration. It has been also found that compound 8c (30 mg kg−1) showed a reduction in the area under the curve from 0 to 120 min [(AUC) 0–120 min] to 37.46%, which was found to be approximately similar to the hypoglycemic profile of alogliptin (standard). The activity of compound 8c was also investigated in STZ-induced diabetic rats where it showed a dose-dependent decrease in blood glucose levels together with an improvement in insulin levels probably via inhibition of DPP-4. The effect of compound 8c was also investigated on the lipid profile and antioxidant enzyme system.
Introduction
Diabetes mellitus (DM) is considered a chronic metabolic disorder that either results from the impaired secretion of insulin from the pancreas or inadequate utilization of the insulin by the body, or both.1 It is considered as a major public health concern across the world, affecting nearly 285 million people and expected to rise to 439 million people by 2030.2 Most of the DM patients are categorized as non-insulin-dependent diabetes mellitus (NIDDM) due to resistance to insulin action, improper secretion of insulin and excessive production of hepatic glucose.3,4 As a result, currently several drugs that are used to maintain the DM condition is often associated with hyperglycemia.5 The efficacy of these drugs has been seriously compromised owing to various side effects, such as hepato-toxicity, abdominal pain, flatulence, diarrhea and hyperglycemia.6 Therefore, the discovery of new antidiabetic agents with improved efficacy has been much sought after to provide better relief. In this regard, the therapy of glucagon-like peptide-1 (GLP-1) has emerged as a promising treatment for type 2 diabetes.7 The incretin hormone, GLP-1, causes stimulation of the biosynthesis and secretion of insulin in a glucose-dependent manner together with suppression of glucagon.8 However, active GLP-1 is rapidly cleaved to its inactive form by the action of dipeptidyl peptidase 4 (DPP-4) resulting in no alteration of insulin levels. Thus, inhibition of DPP-4 offers selective blood glucose lowering effect by increasing the level of active GLP-1 with improved patient compliance, such as a reduction in appetite.9,10Numerous DPP-4 inhibitors are used in clinical practice, including sitagliptin,11 vildagliptin12 and saxagliptin.13 Omarigliptin (MK-3102), a sulfonamide based DPP-4 inhibitor developed by Merck proved to exert potent antidiabetic activity. The pharmacokinetic studies showed that omarigliptin is suitable for once weekly dosing, which makes it unique among the other DPP-4 inhibitors.14 Imeglimin is the first novel class of the –Glimins– belonging to the class of 1,3,5-triazine for the treatment of type 2 diabetes.15 The drug acts via a unique mechanism of action targeting mitochondria bioenergetics, which in-turn improve mitochondrial function, resulting in the restoration of both glucose-dependent insulin secretion and insulin action.16 Moreover, it can also be able to entirely protect the beta cells, or endothelial cells, from death triggered by oxidative stress. 1,3,5-Triazines also showed significant inhibition of DPP-4 activity in vitro.17 In a recent study, it has been found that thiazole derivatives showed DPP-4 inhibitory activity. Moreover, it also exhibits potent antidiabetic activity in the oral glucose tolerance test.18
Based on the distinct and specific pharmacological advantages inherent with sulfonamide, thiazole and 1,3,5-triazine scaffolds, we intended to develop a hybrid skeleton of sulfonamide-1,3,5-triazine–thiazole in a search for novel and potent DPP-4 inhibitors as antidiabetic agent, Fig. 1.
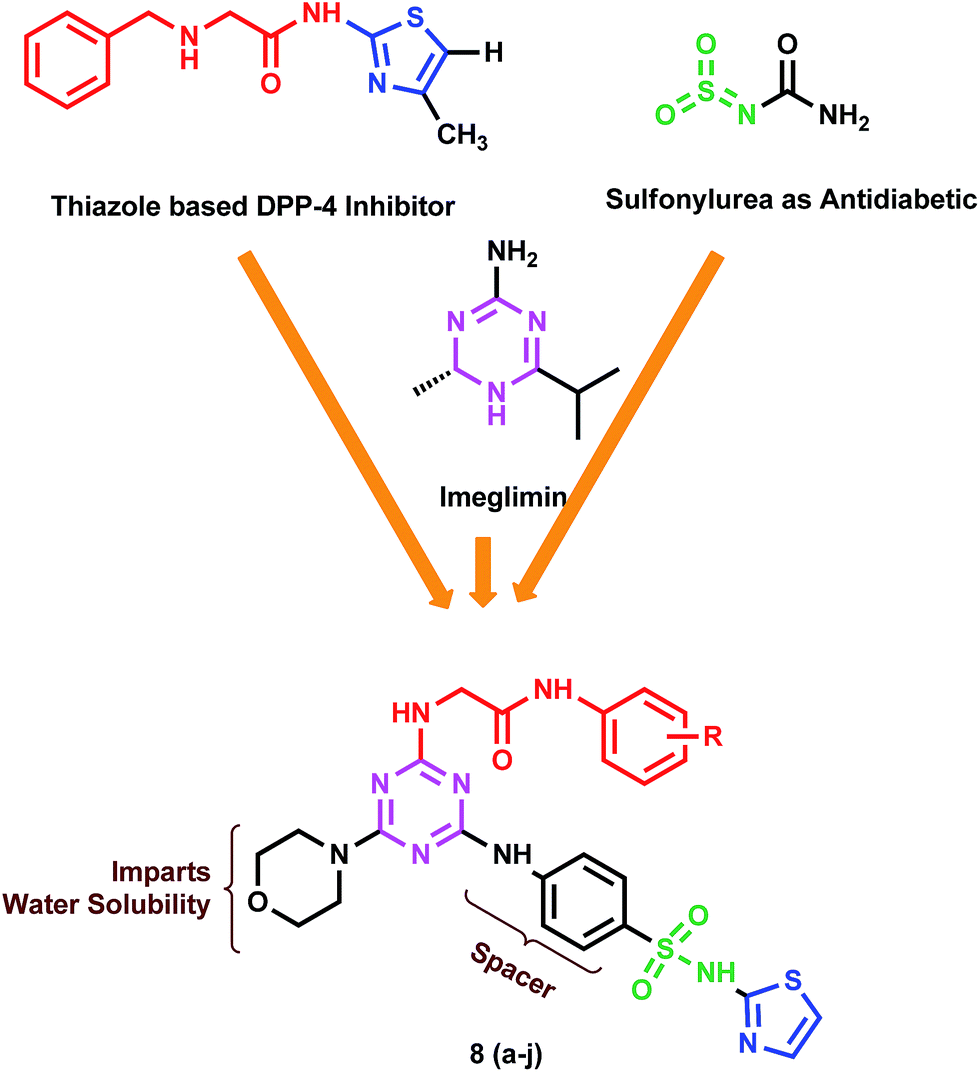 | ||
| Fig. 1 Design strategy of target compounds 8(a–j). | ||
Result and discussion
Chemistry
The synthesis of the target hybrid compounds were achieved in excellent yield via the facile synthetic route shown in Scheme 1. Briefly, the commercially available starting material cyanuric chloride (1) was allowed to react with ammonia gas under 0–5 °C to afford 4,6-dichloro-1,3,5-triazin-2-amine (2). Then, the mono-substituted 1,3,5-triazine derivative was further allowed to react with morpholine at 40–45 °C to afford 4-chloro-6-morpholino-1,3,5-triazin-2-amine (3). The resulting compound was then reacted with 4-amino-N-(thiazol-2-yl)benzenesulfonamide to afford 4-((4-amino-6-morpholino-1,3,5-triazin-2-yl)amino)-N-(thiazol-2-yl)benzenesulfonamide (4). The second part of the synthesis deals with the development of structural fragment needed for the induction of diversity to the core scaffold. This step corresponds to the preparation of N-substituted chloracetamides 7(a–j) in excellent yield by nucleophilic substitution reaction of various distinguished amines 6(a–j) with chloroacetyl chloride (5). The various substituted acetamide derivatives (7) were further allowed to react with the previously synthesized 4-((4-amino-6-morpholino-1,3,5-triazin-2-yl)amino)-N-(thiazol-2-yl)benzenesulfonamide (4) to afford the corresponding final target derivatives 8(a–j) in an efficient manner. The structures of the synthesized target derivatives were ascertained by FT-IR, 1H-NMR, 12C-NMR, mass spectroscopy and elemental analysis. FT-IR spectra of all derivatives 8(a–j) were characterized by the appearance of a strong band at 3383–3398 cm−1 attributed to N–H group. Furthermore, another band at 3078–3092 cm−1 was attributed to the stretching vibrations of the aromatic C–H group. Many strong absorption bands appeared at 1714–1726 cm−1, which confirmed the existence of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group. The band corresponding to the CN triazine ring appears at 1608–1617 cm−1. Moreover, the appearance of a strong band at 1621–1629 cm−1 confirmed the presence of CC aromatic ring groups. The NO2 group of the aromatic ring appears at 1524 cm−1. Compound 8b a exhibited band at 821 cm−1 that attributed to the stretching vibrations of the C–Cl group. The 1H NMR spectra of title compounds 8(a–j) reveal a doublet corresponding to the phenyl ring at 7.47–7.27 ppm. The 1,3,5-triazine NH proton appeared at 3.96 ppm. The resonance due to the morpholine proton was found in the range of 3.67–3.54 ppm. Furthermore, the resonance at 3.16 ppm confirmed the presence of the side chain methylene proton with a single peak of secondary amide appear at 7.23 ppm. The thiazole ring proton appeared as two singlet peaks at 7.26–6.97 ppm. Finally, all structures of the synthesized derivatives 8(a–j) were confirmed by elemental analysis and mass spectroscopy.
O group. The band corresponding to the CN triazine ring appears at 1608–1617 cm−1. Moreover, the appearance of a strong band at 1621–1629 cm−1 confirmed the presence of CC aromatic ring groups. The NO2 group of the aromatic ring appears at 1524 cm−1. Compound 8b a exhibited band at 821 cm−1 that attributed to the stretching vibrations of the C–Cl group. The 1H NMR spectra of title compounds 8(a–j) reveal a doublet corresponding to the phenyl ring at 7.47–7.27 ppm. The 1,3,5-triazine NH proton appeared at 3.96 ppm. The resonance due to the morpholine proton was found in the range of 3.67–3.54 ppm. Furthermore, the resonance at 3.16 ppm confirmed the presence of the side chain methylene proton with a single peak of secondary amide appear at 7.23 ppm. The thiazole ring proton appeared as two singlet peaks at 7.26–6.97 ppm. Finally, all structures of the synthesized derivatives 8(a–j) were confirmed by elemental analysis and mass spectroscopy.
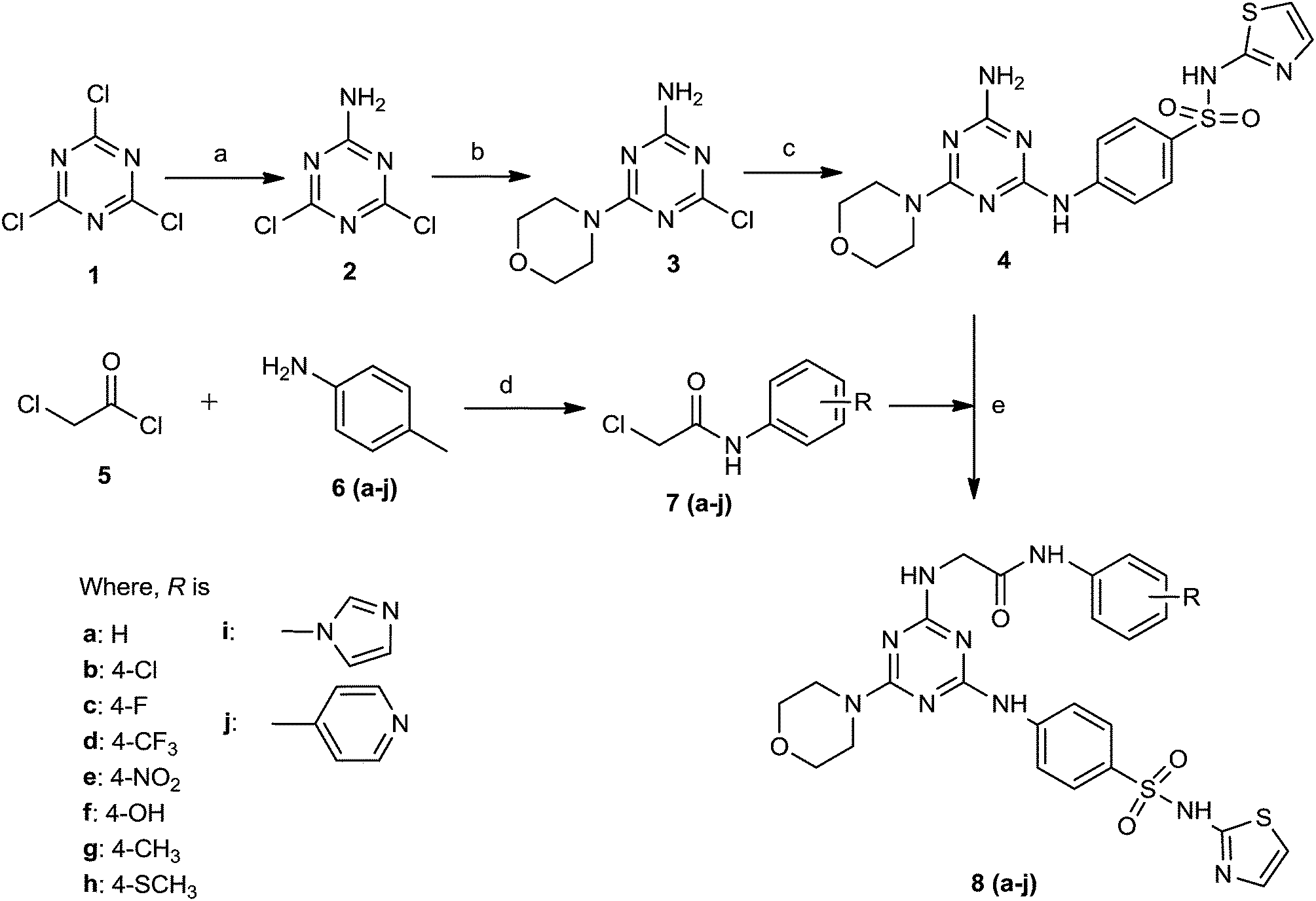 | ||
| Scheme 1 Synthesis of target compounds 8(a–j). Reagent and conditions: (a) NH3, 0–5 °C, 3 h, NaHCO3. (b) Morpholine, 40–45 °C, 4 h, NaHCO3. (c) 4-Amino-N-(thiazol-2-yl)benzenesulfonamide, pyridine, K2CO3, reflux, 10 h. (d) NHR (a–j), triethylamine, 1,4-dioxane, reflux, 6–8 h. (e) Pyridine, 1,4-dioxane, reflux, 9 h. | ||
DPP inhibitory activity
The various synthesized derivatives were assessed for their potential inhibitory activity against numerous DPP enzymes, for instance, DPP-4, DPP-8 and DPP-9, and their inhibitory profiles have been tabulated in Table 1. The compounds were designed in a manner to assess the effect of minor structural variation on the biological activity. The results of the inhibitory data depict that the entire set of compounds showed considerable inhibitory activity against DPP-4 in enzyme-based in vitro assays. Among the tested derivatives, compound 8c was found more potent than alogliptin that was used as a standard. Moreover, as seen in Table 1, none of the compounds showed inhibitory activity against DPP-8 and DPP-9, suggesting that the designed molecules showed selective inhibition of DPP-4 when compared to DPP-8 and DPP-9.| Compound | DPP-4 IC50 (nM) | DPP-8 IC50 (nM) | DPP-9 IC50 (nM) | hERG (μM) |
|---|---|---|---|---|
| a NA: not available. | ||||
| 8a | 544.35 | >25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
>25000 |
NT |
| 8b | 24.03 | >25000 |
>25000 |
NT |
| 8c | 2.32 | >25000 |
>25000 |
505.21 |
| 8d | 18.54 | >25000 |
>25000 |
486.05 |
| 8e | 43.62 | >25000 |
>25000 |
450.62 |
| 8f | 63.32 | >25000 |
>25000 |
410.00 |
| 8g | 145.63 | >25000 |
>25000 |
320.55 |
| 8h | 205.18 | >25000 |
>25000 |
310.14 |
| 8i | 277.33 | >25000 |
>25000 |
250.45 |
| 8j | 392.45 | >25000 |
>25000 |
NT |
| Alogliptin | 3.56 | — | — | >34 |
In the case of DPP-4 inhibition, compound 8a, which has no substituent on the scaffold, showed the least amount of inhibitory activity than the other tested derivatives. Whereas on introducing the p-chloro group, the inhibitory potency of the compound improved significantly (24.03 nM). More surprisingly, the addition of fluoro (8c) in the position of chloro, displayed the most potent inhibition of DPP-4 (2.32 nM) compared to the rest of the analogues. In response to that, the next analogue 8d, was synthesized having a trifluoromethyl to assess the effect of additional fluoro groups. The results showed that introduction of additional fluorine atoms causes a mild drop in activity. The presence of non-halogen electron withdrawing p-nitro (8e) also revealed mild inhibitory activity. In the next instance, the study was aimed to define the effect of electron donating groups on the inhibitory potency (8f, 8g and 8h). Disappointingly, these groups did not influence the activity. As shown in Table 1, compound 8f, 8g and 8h displayed a progressive decline in the activity, exhibiting only moderate inhibition of DPP-4. The rest of the compounds were synthesized to analyze the effect of additional aromaticity on the activity (8i and 8j). The comparative inhibitory results revealed that, compound 8i and 8j showed mild to moderate inhibitory activity, i.e., 277.33 nM and 392.45 nM, respectively.
The structure–activity relationship studies suggest that changes performed in the core skeleton have a profound influence on the inhibitory activity. It has been clearly depicted that compounds containing electron withdrawing groups showed more pronounced inhibitory activity in comparison to the electron donating substituents. Moreover, it has been also found that, the presence of additional aromaticity does not influence the activity. These results suggest that, the receptor pocket is small enough to accommodate only small fragments, not the bigger fragments, because of potential steric clashes.
In accordance with new FDA guidelines, the novel therapies for type 2 DM should be devoid of any cardiovascular risk. Consequently, it is worthwhile to determine the effect of these novel inhibitors on the human Ether-à-go-go Related Gene (hERG) study. Thus, these constitutive compounds were analyzed for their ability to inhibit the hERG ion channel. As shown in Table 1, none of the synthesized derivatives exhibited inhibition of hERG ion channel at the tested dose. Results suggest that, these compounds are potentially devoid of cardiac side-effects.
Docking analysis of compound 8c
In order to explicate the key structural parameters vital for DPP-4 inhibition, a molecular docking of compound 8c was carried out in the active site region of the DPP-4 (2FJP.pdb) enzyme by the CDOCKER module of Discovery Studio 2.5. Numerous structural analyses of the DPP-4 protein model suggested that the enzyme has been broadly classified with two domains, in which one domain is represented by an eight-stranded β-propeller at the N-terminus. Whereas, the other domain consists of a α/β-hydrolase at the C-terminus.Particularly, the binding site of the DPP-4 enzyme has been divided into three specific regions: the S1 pocket, the N-terminal recognition region and the S2 pocket. Docking results showed that ligand 8c was efficiently docked in the active site of the catalytic triad of Ser630, Asp708 and His740, encompassing both the S1 and S2 pockets, shown in Fig. 2. The morpholine fragment of compound 8c was deeply engulfed in the S1 pocket lined by residues like Tyr631, Val656, Trp659, Tyr662, Tyr666 and Val711 that were found adjacent to the catalytic triad of the active site via formation of an H-bond with Ser630. The thiazole sulfonamide fragment of the ligand was found positioned deep in the N-terminal recognition region formed by Glu205, Glu206, and Tyr662 with the formation of two H-bonds with Arg358. It has been also found that the terminal phenyl bearing fluorine interacts with Tyr547 via the formation a non-covalent pi–pi interaction with the S2 pocket. Moreover, the morpholine and 1,3,5-triazine were revealed to create pi–sigma and pi–cation interactions with Arg125 and Tyr662, respectively. In terms of scoring, compound 8c showed CDOCKER interaction energy of 57.80. The results of the docking investigation suggested that compound 8c was able to bind efficiently with all three specific regions (the S1 pocket, the N-terminal recognition region and the S2 pocket) of the DPP-4 enzyme active site and this might be the reason for its excellent inhibitory activity.
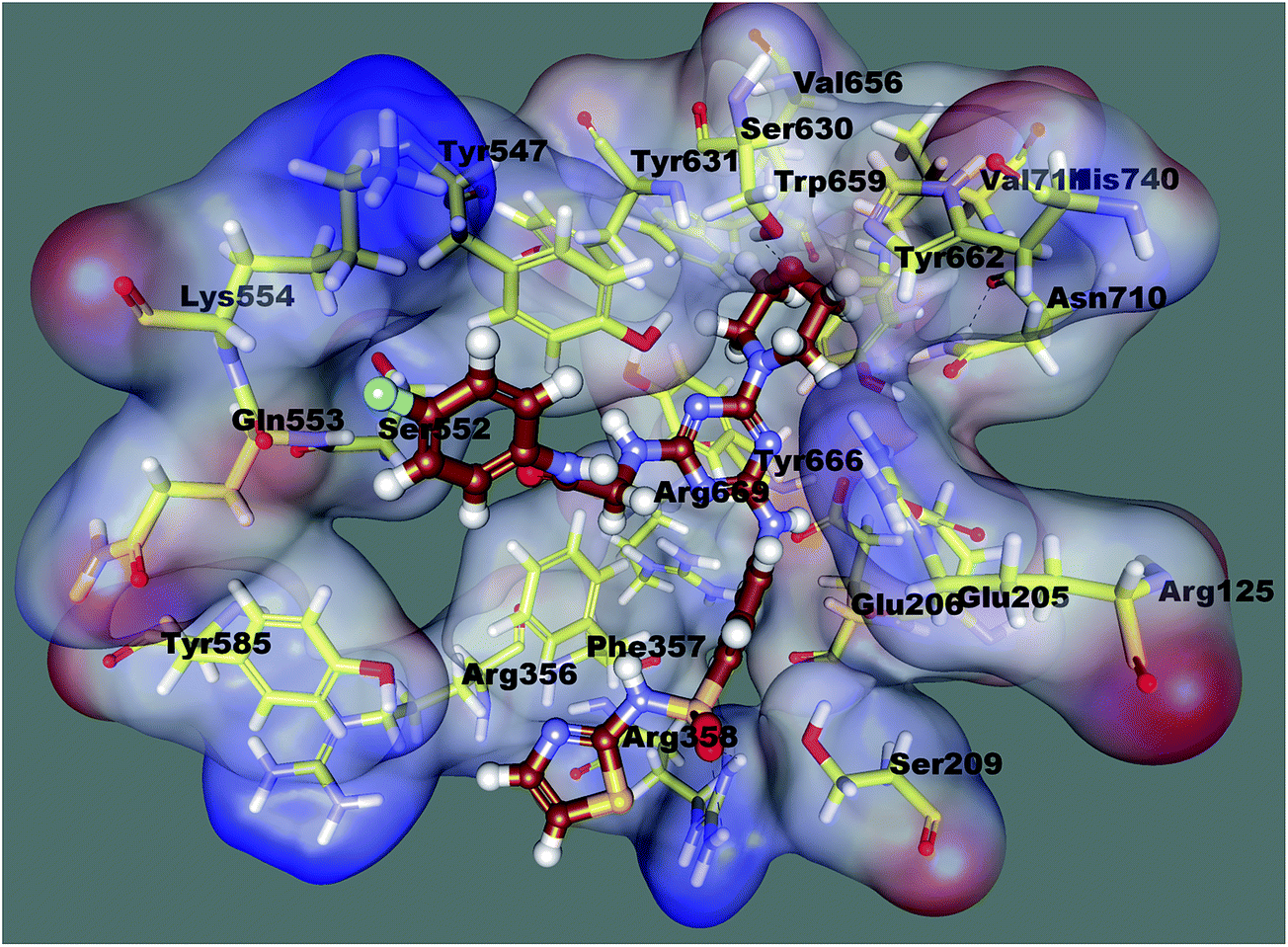 | ||
| Fig. 2 Docked orientation of compound 8c in the active site of the DPP-4 enzyme. | ||
In vivo pharmacological activity of compound 8c
Owing to excellent in vitro potency and selectivity, compound 8c (3, 10 and 30 mg kg−1), alogliptin (3 mg kg−1) or vehicle (0.5% methylcellulose aqueous solution) was orally administered to 12 h-fasted ICR mice (n = 8 in each group) 30 min prior to an oral glucose challenge (2.5 g kg−1). The blood glucose levels of the mice were examined at definite intervals of time, i.e., −30, 0, 30, 60 and 120 min. The results shown in Fig. 3 indicated that compound 8c was found to exhibit concentration-dependent improvement of glucose tolerance in ICR after oral administration. It has been also found that compound 8c (30 mg kg−1) showed a reduction in area under the curve from 0 to 120 min (AUC)0–120 min to 37.46%, which was found to be approximately similar to the hypoglycemic profile of alogliptin (standard).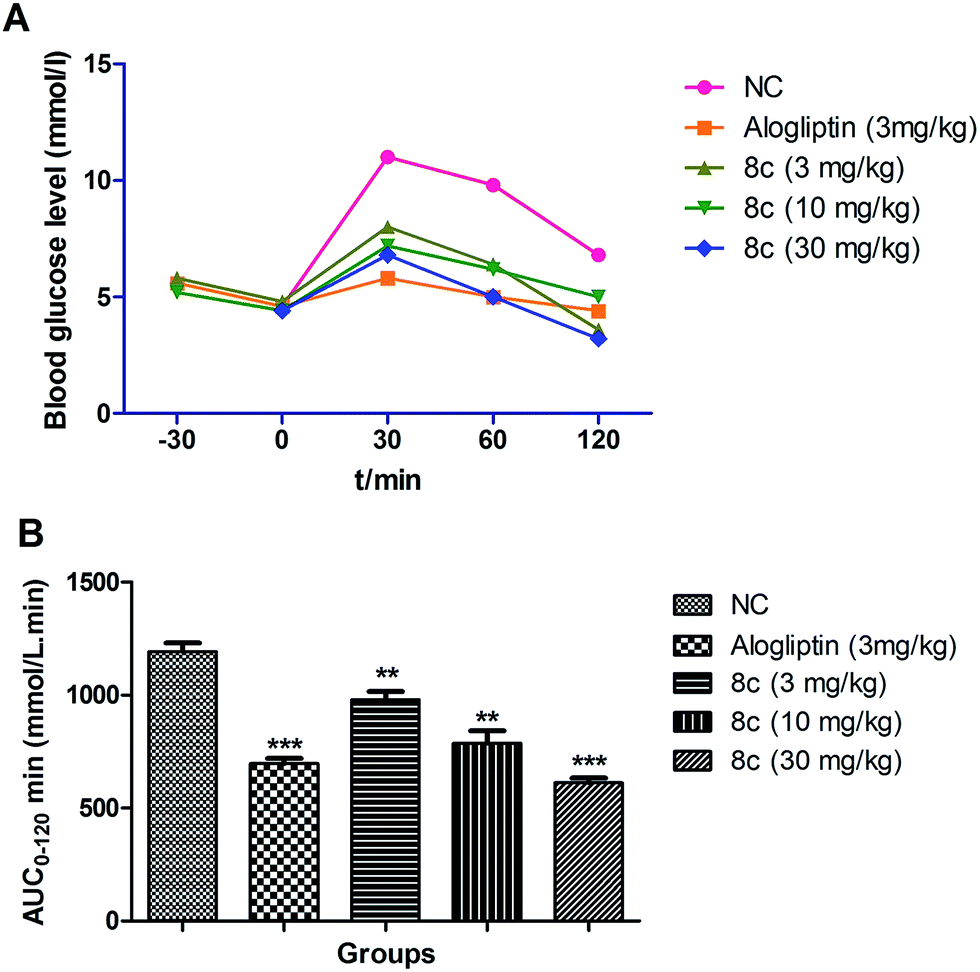 | ||
| Fig. 3 Effect of compound 8c throughout an OGTT in male ICR mice. (A) Time-dependent effect on blood glucose after oral administration of compounds followed by 2.5 g kg−1 oral glucose challenge. Date in (B) represent AUC0–120 min of blood glucose levels. Values are mean ± SEM (n = 8). **P ≤ 0.01 compared to vehicle-treated ICR mice by Student's t test; ***P ≤ 0.001 compared to vehicle-treated ICR mice by Student's t test. | ||
Antidiabetic activity in STZ-induced diabetes in rats
| S. no. | Biochemical parameter | Normal control | Normal control + 8c (30 mg kg−1) | STZ control | STZ + 8c (3 mg kg−1) | STZ + 8c (10 mg kg−1) | STZ + 8c (30 mg kg−1) | STZ + glibenclamide (10 mg kg−1) |
|---|---|---|---|---|---|---|---|---|
| a Values are given as mean ± SEM of six rats in each group. a(P < 0.01) compared with the corresponding value for normal control animals (group I); b(P < 0.01) compared with the corresponding value for diabetic control animals (group II); ns – non significant. | ||||||||
| 1 | Fasting plasma glucose (mg dL−1) | 82.1 ± 0.302 | 77.2 ± 0.365 | 365.3 ± 7.242a | 243.2 ± 4.34b | 187.3 ± 0.434b | 98.3 ± 4.439b | 111 ± 4.584b |
| 2 | Fasting plasma insulin (μU mL−1) | 12.1 ± 2.139 | 12.3 ± 2.344 | 2.8 ± 0.933a | 4.9 ± 0.839b | 7.2 ± 0.382b | 11.2 ± 1.328b | 10.2 ± 1.039b |
| 3 | Hexokinase (μg per mg of tissue) | 154 ± 4.64 | 155.3 ± 5.324 | 80.4 ± 3.103a | 109.3 ± 3.432b | 128 ± 1.839b | 150.2 ± 1.327b | 144.2 ± 2.123b |
| 4 | Glucose-6-phosphatase (unit per mg of tissue) | 8.1 ± 0.831 | 8.2 ± 0.802 | 14.9 ± 0.734a | 12.9 ± 0.839b | 11.8 ± 0.436b | 8.6 ± 0.439b | 9.6 ± 0.435b |
| 5 | Fructose-1-6-biphosphatase (unit per mg of tissue) | 27.2 ± 1.323 | 27.5 ± 1.928 | 59.3 ± 3.435a | 47 ± 1.932b | 40.2 ± 1.738b | 33 ± 0.325b | 35.2 ± 0.324b |
| 6 | Total cholesterol (mg dL−1) | 66.4 ± 3.564 | 67 ± 3.454 | 141.5 ± 2.837a | 130.1 ± 1.932b | 102.1 ± 2.321b | 77.2 ± 3.901b | 84; 3 ± 4.343b |
| 7 | Triglycerides (mg dL−1) | 78.1 ± 2.434 | 78.6 ± 2.839 | 145.1 ± 4.53a | 131.9 ± 2.843b | 111 ± 2.344b | 91.6 ± 3.545b | 98.4 ± 2.323b |
| 8 | Total HDL cholesterol (mg dL−1) | 57.3 ± 2.343 | 57.1 ± 3.874 | 26.1 ± 2.432a | 39.1 ± 2.432b | 45.1 ± 1.343b | 52.1 ± 1.023b | 53.9 ± 1.823b |
| 9 | Total LDL cholesterol (mg dL−1) | 6.8 ± 0.323 | 6.9 ± 0.834 | 87.2 ± 0.383a | 65.2 ± 0.432b | 36.2 ± 0.938b | 8.6 ± 1.928b | 10.8 ± 0.323b |
| 10 | Total VLDL cholesterol (mg dL−1) | 15.8 ± 0.92 | 15.9 ± 0.324 | 29.1 ± 0.326a | 26.4 ± 0.454b | 22.1 ± 0.543b | 18.5 ± 0.982b | 10.8 ± 0.323b |
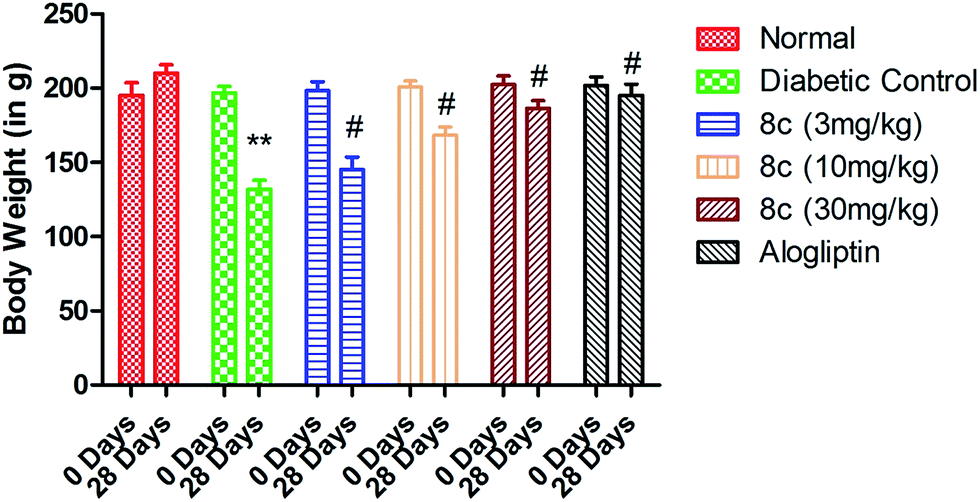 | ||
| Fig. 4 Effect of varying dose of compound 8c on the body weight of the normal and STZ-induced diabetic rats. Values are expressed as mean ± SD (n = 8 rats in each group). Statistical significance was evaluated by one-way analysis of variance (ANOVA). **P < 0.05 as compared with normal rats, #P < 0.05 as compared to diabetic control. | ||
| S. no. | Antioxidant parameters | Normal control | Normal control + 8c (30 mg kg−1) | STZ control | STZ + 8c (3 mg kg−1) | STZ + 8c (10 mg kg−1) | STZ + 8c (30 mg kg−1) | STZ + glibenclamide (10 mg kg−1) |
|---|---|---|---|---|---|---|---|---|
| a Values are given as mean ± SEM of six rats in each group. a(P < 0.01) compared with the corresponding value for normal control animals (group I); b(P < 0.01) compared with the corresponding value for diabetic control animals (group II); ns – non significant. | ||||||||
| 1 | SOD (U per mg of protein) | 7.9 ± 0.432 | 7.8 ± 0.435 | 2.4 ± 0.432a | 4.4 ± 0.783b | 5.4 ± 0.432b | 7.1 ± 0.343b | 6.8 ± 0.432b |
| 2 | CAT (U per mg of protein) | 72.7 ± 2.43 | 70.8 ± 1.43 | 38.6 ± 1.930a | 45.8 ± 1.068b | 53.8 ± 1.158b | 68±1b | 66.1 ± 1.434b |
| 3 | GPx (nmol per mg of protein) | 33.2 ± 1.43 | 32.9 ± 1.432 | 13.8 ± 0.849a | 19.8 ± 0.948b | 24.9 ± 0.434b | 31.7 ± 0.435b | 30.4 ± 0.435b |
| 4 | MDA (nmol per mg of protein) | 0.22 ± 0.014 | 0.24 ± 0.008 | 0.59 ± 0.014a | 0.43 ± 0.011b | 0.34 ± 0.012b | 0.28 ± 0.003b | 0.29 ± 0.006b |
Experimental
Chemicals used in the present study were procured from Sigma Aldrich (USA). The spectra of 1H NMR and 13C NMR were recorded on a Bruker Avance 400 and Bruker Avance 500 spectrophotometer, respectively. The chemical shifts are expressed in parts per million (ppm) and coupling constants are expressed in Hertz (Hz). The low-resolution mass spectra (MS) were recorded on a Waters ZQ LC/MS single quadrupole system equipped with an electrospray ionization (ESI) source. Elemental analyses of the final derivatives were performed using a Vario Elemental analyzer. Thin-layer chromatography was performed on 0.25 mm Merck silica gel plates (60F-254) and visualized under UV light.General procedure for the synthesis of 4,6-dichloro-1,3,5-triazin-2-amine (2)
The synthesis of mono-substituted 1,3,5-triazine was performed in accordance with the earlier reported procedure. The solution of 2,4,6-tri chloro-1,3,5-triazine (1) (0.1 mol) in 25 mL acetone was maintained at the temperature 0–5 °C. Ammonia solution was added constantly to the above solution and stirred for 3 h followed by addition of NaHCO3. The product was filtered and washed with cold water and recrystallized with ethanol to afford pure products.19General procedure for the synthesis of 4-chloro-6-morpholino-1,3,5-triazin-2-amine (3)
4,6-Dichloro-1,3,5-triazin-2-amine (2) (0.1 mol) was added into 50 mL of acetone maintaining the temperature 40–45 °C. Morpholine was added constantly to the above solution and stirred for 4 h followed by the addition of NaHCO3. The product was filtered and washed with cold water and recrystallized with ethanol to afford the corresponding pure product 3.20General procedure for synthesis of 4-((4-amino-6-morpholino-1,3,5-triazin-2-yl)amino)-N-(thiazol-2-yl)benzenesulfonamide (4)
The above synthesized compound 3 (0.1 mol), 4-amino-N-(thiazol-2-yl)benzenesulfonamide (0.1 mol), pyridine (0.1 mol) and K2CO3 (0.1 mol) were taken in a round-bottomed flask containing 100 mL 1,4-dioxane and fitted with a reflux condenser. The content was refluxed for 10 h at 100–120 °C. When the reaction was completed, the resulting solution was poured on crushed ice. The product was filtered off and washed several times with cold distilled water to free it from the chloride. The product was recrystallized with alcohol to furnish 4.General procedure for synthesis of N-substituted chloracetamides 7(a–j)
Different aromatic amines 6(a–j) (0.01 mol) were added in dry 1,4-dioxane (50 mL), maintaining the temperature 0–5 °C. Chloroacetyl chloride (5) (0.02 mol) and triethylamine (0.01 mol) were added with stirring. These different reaction mixtures were further refluxed for 6 to 8 h and excess of solvent then distilled off. The resultant mixtures were poured onto crushed ice to afford compounds 7(a–j).General procedure for the synthesis of final title derivatives 8(a–j)
Solutions of the N-substituted chloracetamides 7(a–j) (0.01 mol) in 1,4-dioxane (5 mL) were added dropwise to a mixture of 4-((4-amino-6-morpholino-1,3,5-triazin-2-yl)amino)-N-(thiazol-2-yl)benzenesulfonamide (4) (0.01 mol), pyridine (0.015 mol) and dioxane (10 mL) with constant stirring at 0–5 °C for 60 min. The temperature was raised to 70–80 °C and further stirred for 9 h. The mixture was then poured in to water (50 mL) and the formed precipitate was filtered, washed with water and recrystallized with alcohol to furnish 8(a–j).O str), 1628 (CC str), 1608 (CN str), 1341 and 1165 (asymmetric and symmetric SO2 str), 1138 (aromatic C–C str), 989 (C–N str), 781 cm−1; 1H-NMR (400 MHz, DMSO, TMS) δ ppm: 8.12 (d, 2H, J = 7.9 Hz, ArH), 7.95 (d, 2H, J = 7.8 Hz, Ar-H), 7.47 (d, 2H, J = 4.7 Hz, Ar-H), 7.27 (d, 2H, J = 4.6 Hz, ArH), 7.26 (s, 1H, thiazole), 7.23 (s, 1H, sec. amide), 7.06 (s, 1H, J = 1.2 Hz, Ar-H), 6.97 (s, 1H, thiazole), 3.96 (s, 3H, aromatic C–NH), 3.76 (s, 2H, methylene), 3.67–3.54 (m, 8H, morpholine); 13C-NMR (100 MHz, CDCl3) δ, ppm: 176.2, 171.8, 168.5, 165.7, 142.2, 138.5, 137.1, 130.2, 129.8, 128.8, 128.1, 121.7, 113.9, 112.2, 66.4, 55.3, 48.7; mass: 568.62 (M + 1); elemental analysis for C24H25N9O4S2: calculated: C, 50.78; H, 4.44; N, 22.21; found: C, 50.79; H, 4.44; N, 22.23.O str), 1629 (CC str), 1612 (CN str), 1345 and 1162 (asymmetric and symmetric SO2 str), 1135 (aromatic C–C str), 992 (C–N str), 821 (C–Cl str) cm−1; 1H-NMR (400 MHz, DMSO, TMS) δ ppm: 8.14 (d, 2H, J = 7.8 Hz, ArH), 7.93 (d, 2H, J = 7.7 Hz, Ar-H), 7.74 (d, 2H, J = 5.4 Hz, Ar-H), 7.42 (d, 2H, J = 5.3 Hz, ArH), 7.22 (s, 1H, thiazole), 7.24 (s, 1H, sec. amide), 6.98 (s, 1H, thiazole), 3.98 (s, 3H, aromatic C–NH), 3.74 (s, 2H, methylene), 3.69–3.52 (m, 8H, morpholine); 13C-NMR (100 MHz, CDCl3) δ ppm: 176.4, 171.8, 168.6, 165.7, 142.2, 137.1, 136.8, 133.4, 130.2, 129.7, 129.1, 120.5, 113.9, 112.2, 66.4, 55.2, 48.7; mass: 603.08 (M + 1); elemental analysis for C24H24ClN9O4S2: calculated: C, 47.88; H, 4.02; N, 20.94; found: C, 47.89; H, 4.03; N, 20.94.O str), 1627 (CC str), 1615 (CN str), 1347 and 1163 (asymmetric and symmetric SO2 str), 1157 (C–F str), 1138 (aromatic C–C str), 989 (C–N str) cm−1; 1H-NMR (400 MHz, DMSO, TMS) δ ppm: 8.12 (d, 2H, J = 7.9 Hz, ArH), 7.92 (d, 2H, J = 7.6 Hz, Ar-H), 7.73 (d, 2H, J = 5.8 Hz, Ar-H), 7.27 (s, 1H, sec. amide), 7.24 (s, 1H, thiazole), 7.08 (d, 2H, J = 5.2 Hz, ArH), 6.96 (s, 1H, thiazole), 3.95 (s, 3H, aromatic C–NH), 3.72 (s, 2H, methylene), 3.67–3.51 (m, 8H, morpholine); 13C-NMR (100 MHz, CDCl3) δ ppm: 176.4, 171.8, 168.6, 165.9, 162.8, 142.2, 137.3, 134.2, 130.4, 129.8, 120.8, 115.8, 113.9, 112.2, 66.4, 55.3, 48.7; mass: 586.64 (M + 1); elemental analysis for C24H24FN9O4S2: calculated: C, 49.22; H, 4.13; N, 21.53; found: C, 49.23; H, 4.12; N, 21.53.O str), 1624 (CC str), 1612 (CN str), 1349 and 1162 (asymmetric and symmetric SO2 str), 1254 (CF3 str), 1141 (aromatic C–C str), 984 (C–N str) cm−1; 1H-NMR (400 MHz, DMSO, TMS) δ ppm: 8.09 (d, 2H, J = 7.8 Hz, ArH), 7.94 (d, 2H, J = 7.6 Hz, Ar-H), 7.56 (d, 2H, J = 5.3 Hz, Ar-H), 7.21 (d, 2H, J = 5.1 Hz, ArH), 7.25 (s, 1H, sec. amide), 7.23 (s, 1H, thiazole), 6.97 (s, 1H, thiazole), 3.97 (s, 3H, aromatic C–NH), 3.74 (s, 2H, methylene), 3.69–3.49 (m, 8H, morpholine); 13C-NMR (100 MHz, CDCl3) δ ppm: 176.3, 171.8, 168.5, 165.8, 142.2, 141.9, 137.2, 132.4, 130.2, 129.8, 125.4, 124.2, 118.9, 113.9, 112.1, 66.4, 55.2, 48.7; mass: 636.65 (M + 1); elemental analysis for C25H24F3N9O4S2: calculated: C, 47.24; H, 3.81; N, 19.83; found: C, 47.25; H, 3.82; N, 19.81.O str), 1627 (CC str), 1614 (CN str), 1524 (NO2 str), 1345 and 1165 (asymmetric and symmetric SO2 str), 1146 (aromatic C–C str), 987 (C–N str) cm−1; 1H-NMR (400 MHz, DMSO, TMS) δ ppm: 8.22 (d, 2H, J = 7.9 Hz, ArH), 8.09 (d, 2H, J = 8.6 Hz, Ar-H), 7.95 (d, 2H, J = 7.6 Hz, Ar-H), 7.35 (d, 2H, J = 8.4 Hz, ArH), 7.27 (s, 1H, sec. amide), 7.21 (s, 1H, thiazole), 6.95 (s, 1H, thiazole), 3.94 (s, 3H, aromatic C–NH), 3.76 (s, 2H, methylene), 3.71–3.49 (m, 8H, morpholine); 13C-NMR (100 MHz, CDCl3) δ ppm: 176.2, 171.8, 168.6, 165.7, 144.6, 143.7, 142.2, 137.1, 130.2, 129.8, 124.2, 119.8, 113.7, 112.1, 66.3, 55.2, 48.7; mass: 613.67 (M + 1); elemental analysis for C24H24N10O6S2: calculated: C, 47.05; H, 3.95; N, 22.86; found: C, 47.06; H, 3.94; N, 22.86.O str), 1621 (CC str), 1617 (CN str), 1348 and 1161 (asymmetric and symmetric SO2 str), 1149 (aromatic C–C str), 992 (C–N str) cm−1; 1H-NMR (400 MHz, DMSO, TMS) δ ppm: 8.14 (d, 2H, J = 7.8 Hz, ArH), 7.97 (d, 2H, J = 7.5 Hz, Ar-H), 7.31 (d, 2H, J = 5.4 Hz, Ar-H), 7.28 (s, 1H, sec. amide), 7.24 (s, 1H, thiazole), 6.98 (s, 1H, thiazole), 6.67 (d, 2H, J = 5.2 Hz, ArH), 5.34 (s, 1H, Ar-OH), 3.97 (s, 3H, aromatic C–NH), 3.78 (s, 2H, methylene), 3.74–3.52 (m, 8H, morpholine); 13C-NMR (100 MHz, CDCl3) δ ppm: 176.2, 171.8, 168.5, 165.7, 154.2, 142.2, 137.1, 130.2, 129.8, 131.2, 123.1, 116.2, 113.9, 112.2, 66.4, 55.2, 48.7; mass: 584.65 (M + 1); elemental analysis for C24H25N9O5S2: calculated: C, 49.39; H, 4.32; N, 21.60; found: C, 49.40; H, 4.31; N, 21.60.O str), 1623 (CC str), 1612 (CN str), 1343 and 1164 (asymmetric and symmetric SO2 str), 1151 (aromatic C–C str), 994 (C–N str) cm−1; 1H-NMR (400 MHz, DMSO, TMS) δ ppm: 8.17 (d, 2H, J = 7.8 Hz, ArH), 7.94 (d, 2H, J = 7.5 Hz, Ar-H), 7.18 (d, 2H, J = 5.1 Hz, Ar-H), 7.26 (s, 1H, sec. amide), 7.26 (s, 1H, thiazole), 7.08 (d, 2H, J = 4.2 Hz, ArH), 6.93 (s, 1H, thiazole), 3.95 (s, 3H, aromatic C–NH), 3.76–3.49 (m, 8H, morpholine), 3.73 (s, 2H, methylene), 2.28 (s, 3H, Ar-CH3); 13C-NMR (100 MHz, CDCl3) δ ppm: 176.2, 171.8, 168.5, 165.7, 142.2, 137.1, 136.9, 135.5, 130.2, 129.6, 129.1, 121.5, 113.8, 112.1, 66.4, 55.2, 48.8, 21.4; mass: 582.68 (M + 1); elemental analysis for C25H27N9O4S2: calculated: C, 51.62; H, 4.68; N, 21.67; found: C, 51.63; H, 4.68; N, 21.65.O str), 1629 (CC str), 1614 (CN str), 1348 and 1161 (asymmetric and symmetric SO2 str), 1154 (aromatic C–C str), 997 (C–N str) cm−1; 1H-NMR (400 MHz, DMSO, TMS) δ ppm: 8.14 (d, 2H, J = 7.9 Hz, ArH), 7.97 (d, 2H, J = 7.6 Hz, Ar-H), 7.34 (d, 2H, J = 5.4 Hz, Ar-H), 7.28 (s, 1H, sec. amide), 7.24 (s, 1H, thiazole), 7.14 (d, 2H, J = 5.2 Hz, ArH), 6.95 (s, 1H, thiazole), 3.97 (s, 3H, aromatic C–NH), 3.74–3.52 (m, 8H, morpholine), 3.72 (s, 2H, methylene), 2.48 (s, 3H, Ar-SCH3); 13C-NMR (100 MHz, CDCl3) δ ppm: 176.2, 171.8, 168.6, 165.8, 142.1, 137.2, 135.2, 134.8, 131.7, 130.2, 129.7, 120.5, 113.9, 112.1, 66.3, 55.2, 48.8, 14.8; mass: 614.73 (M + 1); elemental analysis for C25H27N9O4S3: calculated: C, 48.92; H, 4.43; N, 20.54; found: C, 48.93; H, 4.43; N, 20.53.O str), 1627 (CC str), 1612 (CN str), 1345 and 1164 (asymmetric and symmetric SO2 str), 1157 (aromatic C–C str), 994 (C–N str) cm−1; 1H-NMR (400 MHz, DMSO, TMS) δ ppm: 8.22 (d, 2H, J = 7.9 Hz, ArH), 8.14 (s, 1H, imidazole), 7.94 (d, 2H, J = 7.6 Hz, Ar-H), 7.76 (d, 2H, J = 5.3 Hz, Ar-H), 7.67 (d, 1H, J = 1.9 Hz, imidazole), 7.64 (d, 2H, J = 5.1 Hz, ArH), 7.27 (s, 1H, thiazole), 7.25 (s, 1H, sec. amide), 7.02 (d, 1H, J = 1.4 Hz, imidazole), 6.98 (s, 1H, thiazole), 3.94 (s, 3H, aromatic C–NH), 3.76–3.53 (m, 8H, morpholine), 3.74 (s, 2H, methylene); 13C-NMR (100 MHz, CDCl3) δ ppm: 176.2, 171.8, 168.5, 165.4, 142.1, 138.2, 137.1, 135.5, 132.5, 130.3, 130.1, 129.7, 122.5, 118.3, 117.2, 113.9, 112.1, 66.4, 55.1, 48.9; mass: 634.70 (M + 1); elemental analysis for C27H27N11O4S2: calculated: C, 51.17; H, 4.29; N, 24.31; found: C, 51.18; H, 4.30; N, 24.31.O str), 1623 (CC str), 1615 (CN str), 1343 and 1158 (asymmetric and symmetric SO2 str), 1159 (aromatic C–C str), 991 (C–N str), cm−1; 1H-NMR (400 MHz, DMSO, TMS) δ ppm: 8.62 (d, 2H, J = 4.8 Hz, pyridine), 8.18 (d, 2H, J = 7.9 Hz, ArH), 7.97 (d, 2H, J = 7.5 Hz, Ar-H), 7.71 (d, 2H, J = 4.8 Hz, Ar-H), 7.52 (d, 2H, J = 4.5 Hz, pyridine), 7.48 (d, 2H, J = 4.3 Hz, ArH), 7.28 (s, 1H, thiazole), 7.27 (s, 1H, sec. amide), 6.94 (s, 1H, thiazole), 3.97 (s, 3H, aromatic C–NH), 3.74–3.51 (m, 8H, morpholine), 3.75 (s, 2H, methylene); 13C-NMR (100 MHz, CDCl3) δ ppm: 176.2, 171.8, 168.5, 165.7, 149.6, 147.2, 142.1, 139.1, 137.8, 137.2, 130.1, 129.7, 127.4, 120.7, 119.6, 113.9, 112.2, 66.4, 55.3, 48.7; mass: 645.74 (M + 1); elemental analysis for C29H28N10O4S2: calculated: C, 54.02; H, 4.38; N, 21.73; found: C, 54.03; H, 4.38; N, 21.72.In vitro assays for DPP-4, DPP-8 and DPP-9
Briefly, the DPP-4 enzymes and chemicals were diluted in an assay buffer (50 mM Tris, pH 7.5 and 0.1% bovine serum albumin, pH 7.4). The above mixture was premixed and poured into the 96-well plates. The filled plates were incubated for 10 min at room temperature in the dark to provide a viable condition for enzyme/inhibitor reaction. The assay was started after the addition of 100 mmol L−1 of Gly-Pro-AMC (in assay buffer), which acts as an initiator to induce the enzymatic reaction. The mixture was further incubated for 20 min at room temperature. The liberation of aminomethylcoumarin (AMC) was quantified by a microplate reader after the cleavage of Gly-Pro-AMC at an excitation wavelength of 360 nm and an emission wavelength of 460 nm. The experiments were performed in triplicate and mean IC50 values were calculated by a curve-fitting.DPP-8 and DPP-9 inhibitory assay
Recombinant human DPP-8 (abcam) and DPP-9 (R&D systems) enzyme activity were performed according to manufacturer protocols. Briefly, each enzyme was diluted 1250× to a final volume of 50 μL in assay buffer (100 mM Tris–HCl, 100 mM NaCl, pH 7.8) and added to 96-well flat-bottom microtiter plates, followed by the addition of 10 μL inhibitor and 40 μL substrate (H-Gly-Pro-AMC, final concentration in the assay, 303 μM). The plates were incubated at 37 °C for 10 min. After incubation, fluorescence was measured using a microplate reader using the excitation 380 nm and emission 460 nm.Assay of nonradioactive Rb+ efflux
This analysis was used to determine the ability to inhibit the hERG potassium channel. Briefly, approximately fifty thousand HEK 293 cells were seeded into 96-well culture plates. The plates were incubated for 24 h at 37 °C. After incubation, the medium was then discarded. The plate wells were filled with open channel buffer (198 mL) and 2 mL of test compound solution (stock solution: 30 nM to 300 mM) except for the control wells. The media of the plate was further replaced by a fresh mixture of 198 mL Rb+ load buffer and 2 mL of test compound for incubation for 3 h at 37 °C. The resulting cell layers were then washed quickly in order to remove extracellular Rb+. In the next instance, to activate the hERG channels, the plates were refilled with a mixture of 198 mL channel opening buffer and 2 mL of test compound, except for the control wells. After incubation for 5 min, the supernatant was carefully removed, collected and cells were lysed by the addition of 200 mL of lysis-buffer. The samples were analyzed on the Ion Channel Reader 8000.Oral glucose tolerance test
For this study, briefly, male ICR mice aged 10 weeks (18–22 g) were procured from the institutional animal house facility. The mice were kept under optimal temperature and humidity conditions with a continuous 12 h/12 h light–dark cycle and had free access to laboratory food and water for 1 week before the experimental period. The male ICR mice were fasted overnight (12 h), weighed and randomly categorized into groups (n = 8). Mice were dosed orally with vehicle (0.5% methylcellulose aqueous solution), alogliptin (suspended in vehicle; 3 mg kg−1) or compound 8c (suspended in vehicle; 3, 10 and 30 mg kg−1) at −30 min. Blood samples were collected at −30 and 0 min by tail bleeding for the determination of glucose concentration. After that, glucose (20% aqueous glucose solution, 2.5 g kg−1) was subsequently administered orally (at 0 min). Moreover, additional blood samples were collected at 30, 60 and 120 min after glucose loading for glucose level determinations. Blood glucose was measured by an automatic glucometer.Antidiabetic activity in STZ-induced diabetes
Group I: normal control rats administered vehicle only.
Group II: normal control rats administered 8c (30 mg per kg body weight).
Group III: diabetic control rats administered drinking water alone.
Group IV: tested rats administered 8c (3 mg per kg body weight).
Group V: tested rats administered 8c (10 mg per kg body weight).
Group VI: tested rats administered 8c (30 mg per kg body weight).
Group VII: tested rats administered glibenclamide (10 mg per kg body weight).
Entire groups of rats received different doses of highly active compound 8c and glibenclamide using an intragastric tube once daily for 28 days, continuously. The blood samples of each animal were collected from puncturing the retro-orbital plexus and were later preserved using anticoagulating agents. The blood samples were then centrifuged at 4000 rpm for 15 min and used for analysis of various biochemical parameters. The plasma insulin level was assayed by the radio-immunoassay method. The serum glucose analyses of entire groups of rats were performed by the glucose oxidase-peroxidase (GOD-POD) method using a glucose estimation kit. Other serum estimations were done by spectrophotometric methods using standard kits and following the manufacturer's instructions. The serum TG (triglyceride), TC (total cholesterol), HDL (High Density Lipoprotein) cholesterol were analyzed using a standard kit. While the level of LDL (Low Density Lipoprotein) and VLDL (Very Low Density Lipoprotein) were estimated with the help of the following formulas.
| LDL = TC/1.19 + TG/1.9 − HDL/1.1 − 38 (mg dL−1) |
| VLDL = triglycerides (mg dL−1)/5 |
Estimation of antioxidant enzymes
For the estimation of antioxidant enzymes, livers from all rat groups were successfully removed and made homogenate. The liver homogenate was prepared with ice chilled 10% potassium chloride solution and it was used to measure the levels and activities of superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx) and malondialdehyde (MDA) by the method of Kumar et al.21,22Conclusion
As a concluding remark, it has been corroborated from the present investigation that entire sets of compound 8 showed selective inhibition of DPP-4 over DPP-8 or DPP-9. These compounds were also found devoid of any cardiac over-activity as confirmed by the hERG ion channel activity, which can pose a risk to its drug-likeness ability. The most active compound, 8c, showed in vivo DPP-4 inhibition accompanied with a blood glucose lowering effect in experimental subjects. It was also found to possess a favorable pharmacokinetic profile. It also showed an improvement of blood glucose levels in a dose-dependent manner in STZ-induced diabetic rats via significant improvement of insulin levels and antioxidant enzyme systems. The present study provides a novel series of potent DPP-4 inhibitors.Conflict of interest
Authors declare no conflict of interest.Acknowledgements
This work is supported by The National Natural Science Foundation of China, (No. 81173393); the Natural Science Foundation of Tianjin City (No. 12JCZDJC25500) and the Innovation Team Program from Logistics University of the Chinese People's Armed Police Force (WHTD201310).Notes and references
- WHO, Definition, diagnosis and classification of diabetes mellitus and its complications, Geneva, 1999 Search PubMed.
- D. Mohan, D. Raj, C. S. Shanthirani, M. Datta, N. C. Unwin and A. Kapur, J. Assoc. Physicians India, 2005, 53, 283–287 Search PubMed.
- J. M. Olefsky, R. R. Revers, M. Prince, R. R. Henry, W. T. Garvey, J. A. Scarlett and O. G. Kolterman, Adv. Exp. Med. Biol., 1985, 189, 176–205 CrossRef CAS PubMed.
- J. M. Olefsky and J. J. Nolan, Am. J. Clin. Nutr., 1995, 61, 980S–986S CAS.
- C. M. Clark Jr, Diabetes Care, 1998, 21, C32–C34 CrossRef.
- R. J. Ligthelm, M. Kaiser, J. Vora and J. F. Yale, J. Am. Geriatr. Soc., 2012, 60, 1564 CrossRef PubMed.
- J. J. Wilkins, M. Dubar, B. Sébastien and C. Laveille, J. Clin. Pharmacol., 2014, 54, 267–278 CrossRef CAS PubMed.
- U. Kielgast, J. J. Holst and S. Madsbad, Curr. Diabetes Rev., 2009, 5, 266–275 CrossRef CAS.
- M. Gutniak, C. Orskov, J. J. Holst, B. Ahren and S. Efendic, N. Engl. J. Med., 1992, 326, 1316–1322 CrossRef CAS PubMed.
- D. J. Drucker and M. A. Nauck, Lancet, 2006, 368, 1696–1705 CrossRef CAS.
- P. Aschner, M. S. Kipnes, J. K. Lunceford, M. Sanchez, C. Mickel and D. E. Williams-Herman, Diabetes Care, 2006, 29, 2632–2637 CrossRef CAS PubMed.
- G. Panina, Diabetes, Obes. Metab., 2007, 9, 32–39 CrossRef CAS.
- D. J. Dave, J. Pharmacol. Pharmacother., 2011, 2, 230–235 CrossRef CAS PubMed.
- W. H. Sheu, I. Gantz, M. Chen, S. Suryawanshi, A. Mirza, B. J. Goldstein, K. D. Kaufman and S. S. Engel, Diabetes Care, 2015, 38, 2106–2114 CrossRef PubMed.
- V. Pirags, H. Lebovitz and P. Fouqueray, Diabetes, Obes. Metab., 2012, 14, 852–858 CrossRef CAS PubMed.
- J. Kim, Y. Wei and J. R. Sowers, Circ. Res., 2008, 102, 401–414 CrossRef CAS PubMed.
- J. K. Srivastava, P. Dubey, S. Singh, H. R. Bhat, M. K. Kumawat and U. P. Singh, RSC Adv., 2015, 14095–14102 RSC.
- D. L. Brown and D. Brillon, J. Natl. Med. Assoc., 1999, 91, 389–395 CAS.
- H. R. Bhat, U. P. Singh, A. Thakur, S. K. Ghosh, K. Gogoi, A. Prakash and R. K. Singh, Exp. Parasitol., 2015, 157, 59–67 CrossRef CAS PubMed.
- U. P. Singh, M. Pathak, V. Dubey, H. R. Bhat, P. Gahtori and R. K. Singh, Chem. Biol. Drug Des., 2012, 80, 572–583 CAS.
- V. Kumar, F. Anwar, D. Ahmed, A. Verma, A. Ahmed, Z. A. Damanhouri, V. Mishra, P. W. Ramteke, P. C. Bhatt and M. Mujeeb, BMC Complementary Altern. Med., 2014, 14, 76 CrossRef PubMed.
- V. Kumar, D. Ahmed, F. Anwar, M. Ali and M. Mujeeb, SpringerPlus, 2013, 2, 639 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2016 |