
An enzyme-mediated in situ hydrogel based on polyaspartamide derivatives for localized drug delivery and 3D scaffolds†
Xu Cheng ,
Jia Liu,
Lei Wang,
Ruoli Wang,
Zhilan Liu* and
Renxi Zhuo
,
Jia Liu,
Lei Wang,
Ruoli Wang,
Zhilan Liu* and
Renxi Zhuo
Key Laboratory of Biomedical Polymers, Ministry of Education, College of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, P. R. China. E-mail: liuzl@whu.edu.cn; Tel: +86-27-68755200
First published on 11th October 2016
Abstract
An injectable and biodegradable enzymatically crosslinked hydrogel based on poly(α,β-aspartic acid) modified with tyramine (TA) and 3-amino-1-propanol (AP) (PASP-g-TA/AP) was prepared under physiological conditions in the presence of horseradish peroxidase (HRP) and hydrogen peroxide (H2O2). The polymer PASP-g-TA/AP was readily synthesized through aminolysis of polysuccinimide (PSI) with TA and subsequently with AP. The gelation time could be tuned from 10 s to 8 min by modulating the concentrations of polymer, HRP and H2O2 cooperatively. Studies on swelling ratio, hydrogel morphology and a rheology analysis further demonstrated the relationship between the crosslinking density and the concentrations of the polymer, HRP and H2O2. The PASP-g-TA/AP hydrogels displayed a considerable capacity to control the release of hydrophilic small model drugs at higher polymer concentrations. The cell culture study showed that COS-7 cells exhibited high viability in vitro and notable proliferation when cultured within the PASP-g-TA/AP hydrogel. After subcutaneous injection into mice, the PASP-g-TA/AP hydrogels formed rapidly in situ and persisted for up to 30 days with an acceptable inflammatory response. These results suggest that the newly described PASP-g-TA/AP hydrogels have good biocompatibility and are promising biomaterials as targeted drug delivery carriers and three-dimensional scaffolds.
1 Introduction
Over the past several decades, hydrogels have been widely studied as biomaterials in the fields of drug delivery1–5 and tissue engineering.6–9 Besides, the application of hydrogels in diagnostics,10 separation of biomolecules or cells and three-dimensional (3D) cell cultivation have also been reported.11–13 This is due to their high water content, great biocompatibility, and beneficial 3D microenvironment that resembles the native extracellular matrix (ECM). Recently, compared to traditional preformed hydrogels, injectable in situ-forming hydrogels have received increasing attention for their limited surgical invasion and facile incorporation of bioactive moieties. Therapeutic agents including cells can be simply mixed with the precursor solution before gelation and then injected into targeted tissue to fill irregular defect sites. Due to these remarkable characteristics, injectable hydrogels have been widely considered as promising materials for 3D cell cultivation and tissue engineering.14,15Injectable hydrogels can be fabricated through physical interactions and chemical reactions. The physical hydrogels cross-linked by the non-covalent interactions always respond to external stimuli including temperature, pH, ionic strength, etc.16–21 They are usually reversible in aqueous solutions and avoid damaging the encapsulated guest molecules during crosslinking. Many of them, however, are weak in vivo and do not exhibit enough long-term stability in tissues, which has largely limited their applications in biological fields. Compared to physical hydrogels, the injectable hydrogels based on chemical reactions such as Michael addition reaction,22,23 photo-crosslinking,24 Schiff-base formation,25,26 boron–catechol interactions,27,28 click reactions29,30 provide tunable stability and functionality. Nevertheless, most of them still have some disadvantages including the difficulty to biodegrade, not biological orthogonal reaction31,32 and the usage of noxious initiators and organic solvents.33 Enzyme-mediated in situ crosslinking hydrogels (typically the redox system of horseradish peroxidase (HRP) and hydrogen peroxide (H2O2)) have captured increasing attention because of their high specificity and little cytotoxicity of HRP. In addition, the gelation time of the system can be well controlled under mild physiological conditions, which is of great significance in practical applications. In the presence of H2O2, HRP effectively catalyzes the oxidative coupling of aniline or phenol derivatives of the polymers to form gel.34,35
To date, enzymatically crosslinked in situ hydrogels based on both natural and synthetic polymers have been reported. Natural polymers such as chitosan,36 dextran,37 xylan38 and hyaluronic acid39 derivatives have been tried to prepare injectable hydrogels in the presence of HRP-based catalysts for tissue regenerative medicine and the treatment of arthritic cartilage or cartilage defects. It is known that the natural polymers have great biocompatibility, but they usually exhibit poor availability and instability in vivo after forming hydrogel. In addition, the arduous purification and the uncontrollability of molecular weight and its distribution are also the main obstacles in their application. In contrast, synthetic polymer hydrogels overcome the drawbacks of natural ones, but their insufficient biocompatibility, the hardness of degradation and the adverse effect on tissue caused by degradation products are the defects that need to be considered in bio-related applications.
In view of the above, synthetic poly(amino acid)s is expected to offer many advantages such as chemical versatility and well-controlled molecular weights,40 more importantly, the desirable biocompatibility and biodegradability. For instance, Ren41 prepared enzymatically crosslinked hydrogels based on a poly(L-glutamic acid) graft copolymer. The hydrogel exhibited good cytocompatibility in vitro and acceptable biocompatibility in vivo. However, the synthesis of the poly(L-glutamic acid)-graft-tyramine/poly(ethylene glycol) (PLG-g-TA/PEG) conjugates suffered from complicated process steps.
Polysuccinimide (PSI), which can be prepared by thermal bulk polymerization of L-aspartic acid facilely, is used for preparing poly(aspartic acid) derivatives and has been intensively studied as hydrogels. According to previous literatures, the molecular weight and distribution of PSI can be well controlled by varying reaction conditions.42 PSI can be easily modified with various functional groups by ring-opening reaction using nucleophilic amine compounds and most of the PSI derivatives exhibit good biomedical properties. Until now, reports on injectable hydrogel based on PSI are yet limited. Lu43 synthesized double components of polyaspartylhydrazide (PAHy) and acetaldehyde-modified poly(2-hydroxyethyl-aspartamide) (PAAld) as precursors of injectable hydrogel. The hydrogel showed excellent drug release profile and low cytotoxicity in vitro, whereas the tunable range of gelation time was not wide due to the limitation of the high viscosity of the solution and poor solubility of PAAld. Gyarmati44 immobilized thiol groups onto poly(aspartic acid) (PASP) as injectable polymer which can form hydrogels in situ. The chemical stability of the polymer is appropriate and gelation time can be tuned between 2–6 min by varying the concentration of the oxidising agent, making the polymer suitable for injectable material. But another oxidising agent instead of sodium bromate may be chosen to improve the biocompatibility for further application. Besides, Fiorica45 prepared in situ hydrogel based on α,β-poly(N-hydroxyethyl)-DL-aspartamide (PHEA) derivatives and hyaluronic acid derivatives grafted by α-elastin. The hydrogel was formed via Michael-type addition under mild conditions. Except for a good resistance towards hydrolytic and enzymatic degradation, the hydrogel showed nice biocompatible for chondrocytes culturing within it that represented potentiality for cartilage tissue engineering.
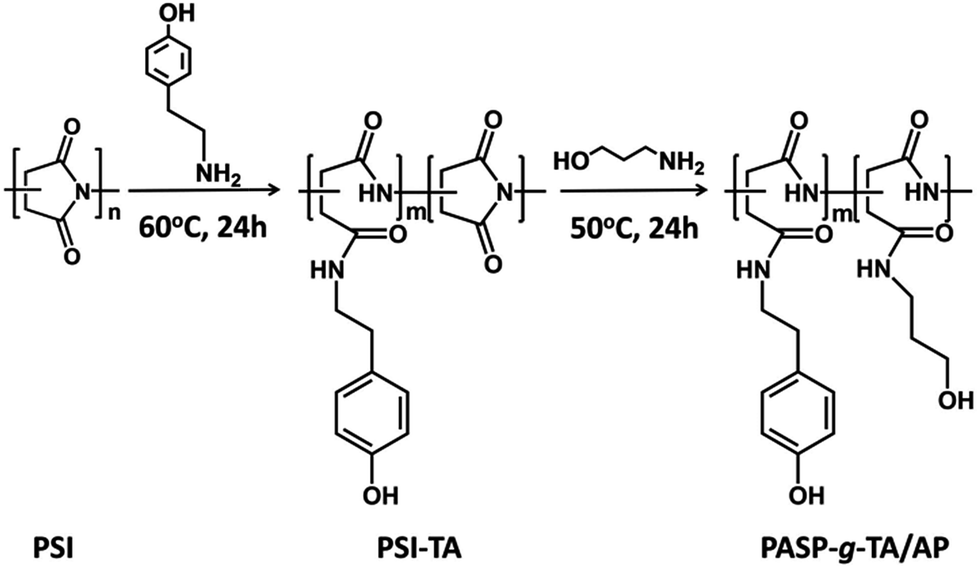
In this paper, an injectable, biocompatible and biodegradable enzymatically cross-linked hydrogel based on PSI was developed for localized drug delivery and 3D scaffolds. As reported, poly(aspartic acid) modified by tyrosine, tyramine (TA) and aminophenol with high substitution degrees could be crosslinked to form gels in the presence of peroxidase/H2O2.46 Due to the low molecular weight of the precursor, the hydrogel formed at high polymer concentration. The electrostatic interaction between the acid groups on the main chain and the active site of peroxidase may have adverse effect on crosslinking. It is also found that α,β-poly(N-3-hydroxypropyl)-DL-aspartamide (PHPA), derived by coupling PSI with 3-amino-1-propanol (AP), has been studied as gene vector and drug carrier because of its excellent biocompatibility and non-toxicity. The characteristics of easy modification and pH-depended biodegradation also provide ideal advantages for in vivo application.47,48 Here, the poly(aspartic acid)-graft-tyramine/3-amino-1-propanol (PASP-g-TA/AP) was synthesized by ring-opening of PSI with TA and subsequently with AP for the aminolysis of the remaining imide ring. For the reason that substitution degree of TA is crucial for the crosslinking density of the hydrogel, a very simple synthetic route was provided in this paper that the substitution degree could be well adjusted by a two-step reaction in one system. The chemical structure of the PASP-g-TA/AP was characterized by 1H NMR. The morphology, gelation time, viscoelastic property, swelling ratio and drug release behavior in vitro of the hydrogels formed with different concentrations of polymer, HRP and H2O2 were investigated. The in vitro cytotoxicity of the gels and cell growth behaviors inside the hydrogel were also studied. Moreover, the tissue biocompatibility in vivo was further investigated by subcutaneous injection of the materials using KM mice as the model animals (Scheme 1). These results will evaluate the potential application of the PASP-g-TA/AP hydrogel in drug delivery, 3D cell cultivation and minimally invasive in situ tissue repair.
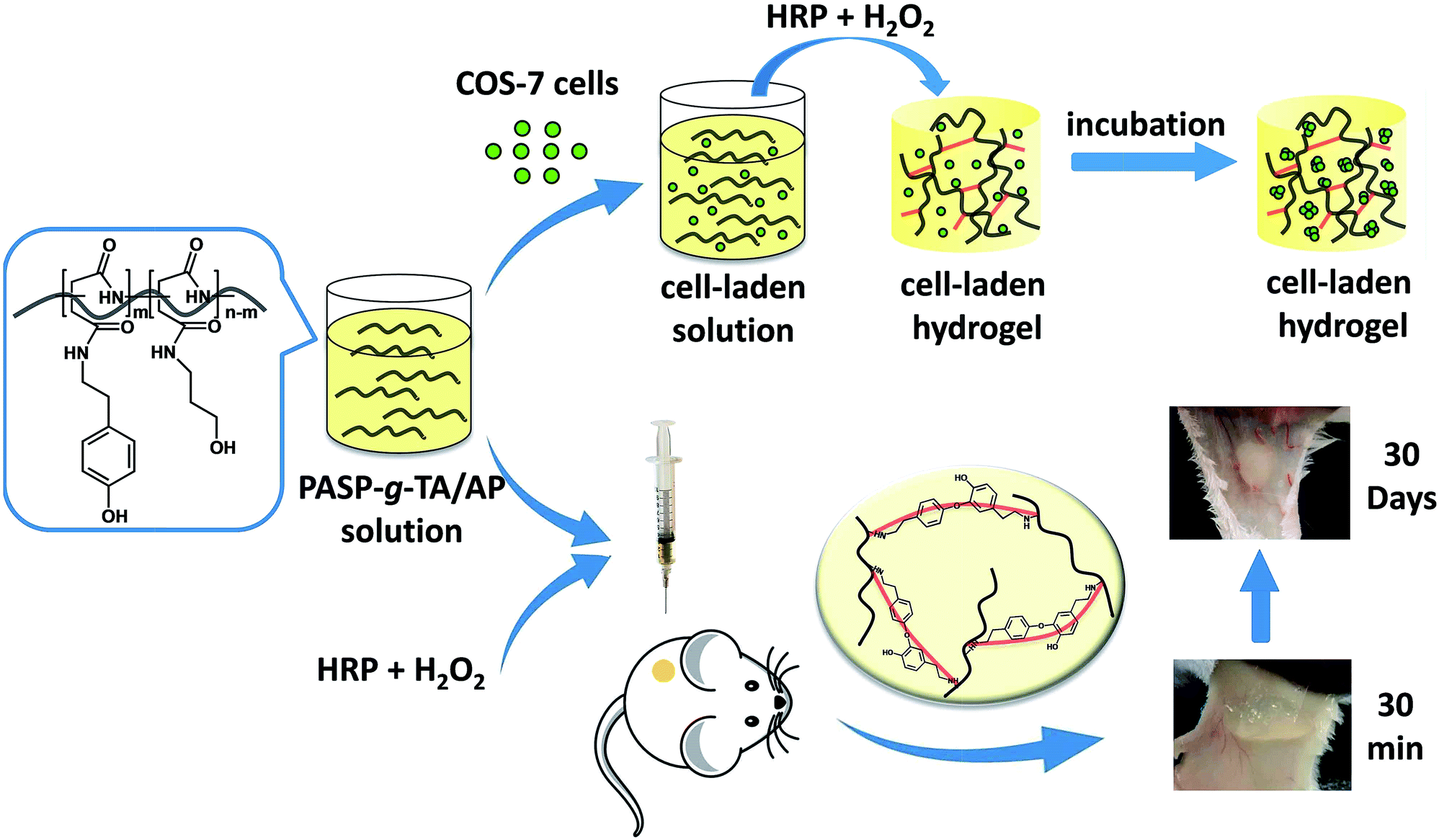 | ||
| Scheme 1 Illustrations of the precursor structure, the process of encapsulation, proliferation of COS-7 cells in vitro and hydrogel formation in vivo. | ||
2 Experimental section
2.1 Materials
L-Aspartic acid, 3-amino-1-propanol (AP), N,N-dimethylformamide (DMF), ortho-phosphoric acid (98%), hydrogen peroxide (H2O2, 30 wt% in H2O), rhodamine B (Rh B) were purchased from Sinopharm Group (China). Tyramine (TA) was purchased from Energy Chemical Company. Horseradish peroxidase (HRP, 250–330 units per mg) was obtained from Sigma Chemical Company. Acridine orange (AO) and ethidium bromide (EB) were purchased from Beijing Dingguo Changsheng Biological Co., Ltd. 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) was purchased from Beijing Junyao Biological Company. Cell Counting Kit-8 (CCK-8) was purchased from Biosharp. All the chemical reagents were of analytical grade and used without further purification.2.2 Characterization
The 1H NMR spectra were measured with a Mercury VX-300 spectrometer at 300 MHz using D2O or DMSO-d6 as solvent. The molecular weights and the molecular weight distributions were determined by a size-exclusion chromatography-multiangle laser light scattering (SEC-MALLS) system consisting of a Waters 2690D separations module, a Waters 2410 refractive index detector, and a Wyatt DAWN EOS MALLS detector. Two chromatographic columns (Styragel HR3 and HR4) were used in series. DMF containing 10 mM of LiBr was used as the mobile phase at a flow rate of 0.3 mL min−1 at 30 °C. The data were analyzed with Astra software (Wyatt Technology). The morphology of the hydrogels was measured by Quanta 200 scanning electron microscope (SEM; FEI instrument, USA). UV-Vis spectra (200–800 nm) were determined by Lambda Bio 40 UV/VIS spectrometer (Perkin-Elmer) at room temperature. Rheological experiments were performed on a controlled stress rheometer (HAAKE Rheo Stress 6000, Thermo Scientific, Germany) equipped with thermostatic water bath. The cell morphology was observed by a confocal laser scanning microscopy (CLSM, Nikon C1si, Japan).2.3 Animals
KM mice (3–4 weeks old, 18–20 g, male) were purchased from Biossci Biotechnology Company (Wuhan, China). The care and use of animals were performed in compliance with Hubei Province Administration Rule of Laboratory Animal. The protocol was approved by the Ethics Committee for Animal Research, Wuhan University, China.2.4 Synthesis of PASP-g-TA/AP
PASP-g-TA/AP was synthesized from PSI, and PSI was synthesized according to the literature.49 Briefly, 30.0 g L-aspartic acid and 15.0 g 98% ortho-phosphoric acid were mixed in a 1 L round-bottomed flask and stirred under reduced pressure in an oil bath at 180 °C for 2.5 h. The reaction mixture was dissolved completely in 200 mL of DMF at 50 °C. Then, the solution was precipitated by an excess of deionized water and washed with deionized water until neutrality. The product was finally dried in a vacuum oven at 110 °C for 24 h and obtained 22.35 g of PSI. TA (1.14 g, 8.29 mmol) was added to PSI (4.02 g, 41.44 mmol of repeat unit) solution in 40 mL of DMF, the mixture was stirred at 60 °C under nitrogen atmosphere. After 24 h, AP (4.98 g, 66.31 mmol) was added and stirred at 50 °C under nitrogen atmosphere overnight.50 The final solution was dialysed against deionized water for 3 days through a dialysis membrane (MWCO, 3.5 kDa). 5.94 g of PASP-g-TA/AP was collected by lyophilization with a yield of 78%.2.5 Hydrogel formation and gelation time determination
The gelation time of the PASP-g-TA/AP hydrogel was determined using the vial-tilting method.51,52 To 200 μL of a polymer solution, freshly prepared HRP solution (50 μL) and H2O2 solution (50 μL) of different concentrations were added and gently mixed. The final concentrations of PASP-g-TA/AP were fixed at 4.1% and 16.7% (w/w) standing for lower and higher concentrations respectively in the following experiments. All solutions used were dissolved in PBS (pH 7.4, 0.01 M). The gel status was identified when no flow was visually observed within 30 s after inverting the vial.2.6 Morphology of the hydrogels
Morphology of PASP-g-TA/AP hydrogel was observed by SEM. The hydrogel samples were kept overnight after formation, then immersed quickly in liquid nitrogen and freeze-dried at −45 °C for 2 days. Lyophilized gels were gold-coated and then viewed by SEM with an accelerating voltage of 15 kV.2.7 Rheological experiments
To evaluate the rheological property of PASP-g-TA/AP hydrogel, 600 μL of the mixture solution containing different concentrations of polymer, HRP, and H2O2 in PBS were quickly transferred to a parallel plate (plate diameter = 40 mm, fixed gap = 0.11 mm) of the rheometer. Silicon oil was placed around the sample to prevent the evaporation of water. The experiments were performed with a frequency of 1 Hz and 1% strain in oscillatory mode at 37 °C. The storage modulus (G′) and loss modulus (G′′) during 30 min was recorded as a function of time.2.8 Swelling ratio
The swelling property of PASP-g-TA/AP hydrogel at 37 °C was measured. The gels freeze-dried for 2 days and dried in vacuum for 24 h were defined as dry gels. The dry gels were immersed in PBS and the swollen gels were obtained. And then the swollen gels were taken out and weighed after removing adhering water on the surface of the hydrogel at predetermined time intervals. The swelling ratio (mean ± SD, n = 4) of the hydrogel was calculated by the following equation:where Ws and W0 are the weights of swollen and dry gel, respectively.
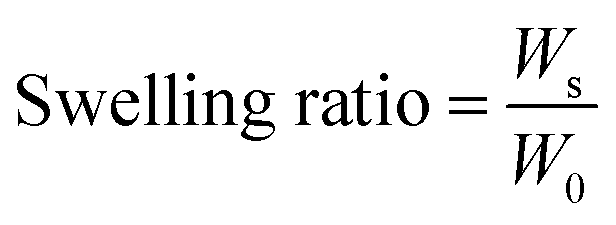
2.9 In vitro drug release behavior
Rh B was used as a model drug for the drug release experiment. 12.5 mg and 50 mg of polymer were respectively dissolved in 200 μL PBS containing of 0.3% (w/w) Rh B. To each sample, freshly prepared HRP solution (50 μL) and H2O2 solution (50 μL) at predetermined concentrations were added. The solutions were agitated gently and thoroughly to form hydrogels and incubated in a 37 °C water bath overnight. The drug loading hydrogel was immersed in 6 mL (V0) PBS buffer and the mixture was placed in a shaking water bath at 37 °C with shaking rate at 60 rpm in the dark. At predetermined intervals, 3 mL (Ve) of release medium was removed for analysis and replaced by an equal volume of fresh buffer. The concentration of released Rh B from hydrogel was quantified by UV spectrophotometer at 554 nm. The cumulative Rh B released percent was calculated using the equation:where Ci represents the concentration of Rh B in the ith sample, and m0 is the gross amount of Rh B in the initial hydrogel.
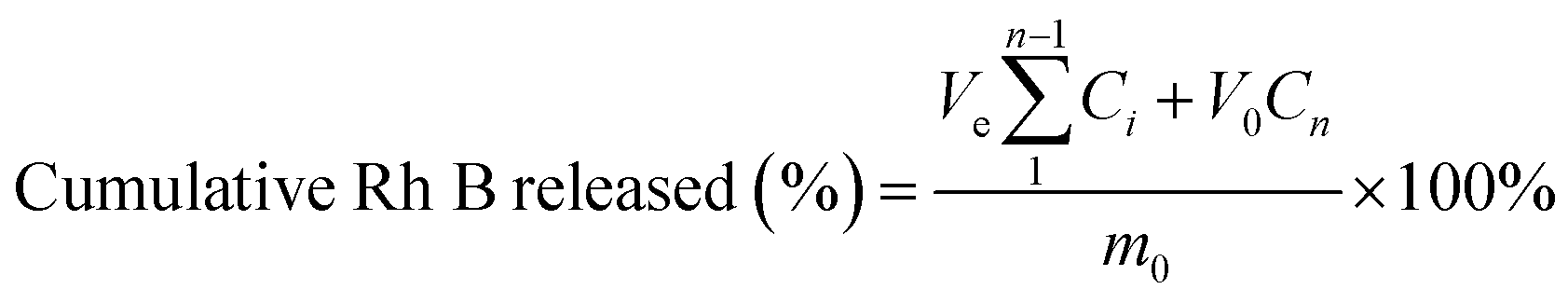
2.10 In vitro cytotoxicity
In vitro cytotoxicity of the PASP-g-TA/AP hydrogel against COS-7 cells was evaluated by MTT assay using a direct contact method between the hydrogel and the cells according to the literature.53,54 Briefly, the precursor solution ([polymer] = 4.1%, [HRP] = 0.016 mg mL−1, [H2O2] = 4.41 mM) was kept overnight after forming gel and cut into 3.2 mm diameter disks with different thickness. The hydrogel disks were sterilized by immersing in 75% alcohol for 2 h and then washed three times with PBS and Dulbecco's modified Eagle's medium (DMEM) (containing 10% fetal bovine serum (FBS), 100 U mL−1 penicillin and 100 μg mL−1 streptomycin) for 30 min for each. COS-7 cells were seeded in a 48-well tissue culture polystyrene (TCPS, Costar) plate at a density of 1 × 104 per well. After incubating at 37 °C under a humidified atmosphere of 95% air and 5% CO2 for 24 h, the medium was replaced with 500 μL per well of fresh medium and the hydrogel disks were added. After incubating for 48 h, the culture medium was replaced with 500 μL per well of fresh medium. 40 μL of MTT (5.0 mg mL−1) solution was added to each well according to the manufacturer's instructions and further incubated for 4 h. 300 μL per well of DMSO was added to dissolve the resulted formazan crystals after removing the medium. The optical density OD values at 570 nm were then measured after removing the hydrogel disks using Microplate Reader model 550 (BIO-RAD, USA). The cell viability was determined by comparing the absorbance of the wells containing hydrogels with that of control wells containing medium only. The final data were shown as the mean value of four replicates plus a standard deviation (±SD) for each sample.The cytotoxicity of the extract of PASP-g-TA/AP hydrogel was also evaluated by measuring the cell viability in the presence of the extract solution. Briefly, 300 μL of hydrogel formed with the same concentrations of polymer, HRP and H2O2 as mentioned in the previous paragraph was immersed in 2 mL PBS and kept in the dark for 7 days to get the extract solution. COS-7 cells were seeded in a 96-well plate at a density of 5 × 103 cells per well. After incubating at 37 °C under a humidified atmosphere of 95% air and 5% CO2 for 24 h, the culture medium per well was replaced by 200 μL of DMEM containing the extract solution. After incubating for 48 h, the DMEM containing extract solution was replaced with 200 μL of fresh medium. Then 20 μL of MTT (5.0 mg mL−1) solution was added to each well and incubated for another 4 h. After removing the medium, 150 μL of DMSO was added. The optical density OD values at 570 nm were then measured with Microplate Reader model 550 (BIO-RAD, U.S.A.). The viable rate (mean ± SD, n = 4) was calculated by the following equation:
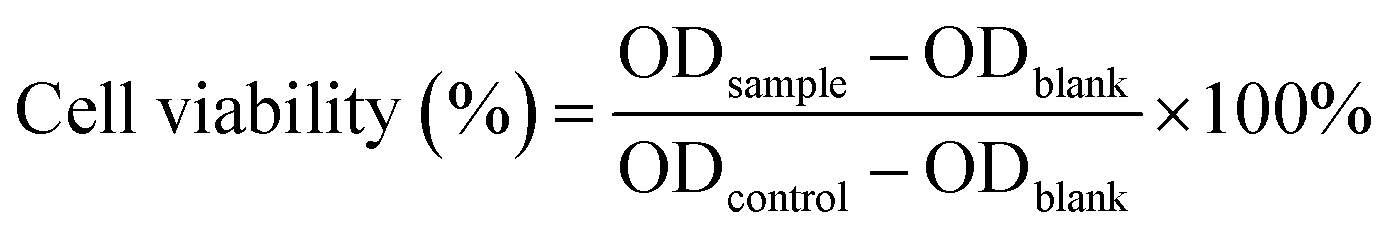
2.11 3D cell culture within PASP-g-TA/AP hydrogels
The lyophilized PASP-g-TA/AP samples were sterilized under UV irradiation for two hours and then dissolved in sterilized PBS. HRP stock solution (0.10 mg mL−1) and H2O2 stock solution (0.09%) freshly prepared in PBS were sterilized by filtration through filters (pore size of 0.22 μm). The hydrogel was then formed in the presence of cells. Briefly, 100 μL of polymer solution containing 12.5 mg of PASP-g-TA/AP was mixed with 100 μL of medium containing harvested cells (1 × 105 cells). To the cell/polymer suspensions, 50 μL of the stock HRP solution and 50 μL of the stock H2O2 solution were added and gently mixed. The mixture was quickly transferred on to a Laser confocal culture dish before gelation. 1 mL of DMEM was added and the hydrogel was incubated at 37 °C under a humidified atmosphere of 95% air and 5% CO2. The viability of encapsulated COS-7 cells following 1, 3 and 6 days of incubation within the hydrogel was observed by CLSM after adding 6 μL of AO (100 μg mL−1) and 6 μL of EB (100 μg mL−1) and incubation for another 1 h. The medium was replaced every day.The proliferation of COS-7 cells within the hydrogel was assessed by CCK-8 assay. 1 mL COS-7 cell suspensions were mixed with 1 mL of PBS containing 125 mg of PASP-g-TA/AP. 500 μL of HRP stock solution and 500 μL of H2O2 stock solution were added and agitated to make a final cell density of 3.3 × 105 mL−1. 64 μL of the mixture solution was quickly transferred to each well in 96-well plate before gelation. 100 μL of culture medium was added on each gel and then the cells were incubated at 37 °C in an atmosphere with 95% humidity and 5% CO2. The medium was replaced every day. At predetermined time, 10 μL of CCK-8 solution (10% v/v in culture medium) was added into each well of the plate. After 4 h incubation, the cell viability in hydrogel was determined by the absorbance values at 450 nm measured using a microplate reader. The absorbance determined with PASP-g-TA/AP hydrogel without cells was used as blank. For the purpose of comparing the cell proliferation cultured in the hydrogels to that cultured on the TCPS, 1 mL COS-7 cell suspensions were mixed with 2 mL PBS to make the same cell density and seeded in 96-well plate. The proliferation was assessed in the same time intervals. The final data were shown as the mean value of four replicates plus a standard deviation (±SD) for each sample.
2.12 In vivo gel formation and biocompatibility
The in vivo gelation and histological reaction of the gel in KM mice were assessed by subcutaneously injection of the materials. For this study, 210 μL of a mixed sterilized solution containing PASP-g-TA/AP, HRP and H2O2 was subcutaneously injected into the back of each mouse before gelation. The concentrations of PASP-g-TA/AP, HRP and H2O2 were the same as those used in proliferation study. At predetermined time intervals, the mice were sacrificed. The gel-implants and the surrounding tissues were excised and fixed in 4% paraformaldehyde buffer solution. All the fixed samples were then embedded in paraffin wax and thin sections (4 μm) were stained with hematoxylin and eosin (H&E). The histological reactions were evaluated by observing the sections using an Olympus BX 50 microscopy (Olympus, Hamburg, Germany).3 Results and discussion
3.1 Synthesis and characterization of PSI and PASP-g-TA/AP
PSI was prepared through acid-catalysed thermal polycondensation of aspartic acid at 180 °C for 2.5 h. The chemical structure of PSI was confirmed by 1H NMR in DMSO-d6 (see ESI Fig. S1†). The characteristic peaks of the succinimide repeating units of PSI appear at 5.26 ppm attributable to methine protons, 2.72 ppm and 3.21 ppm attributable to methylene protons.As the synthesis route shown in Scheme 2, PASP-g-TA/AP was synthesized by the aminolysis of PSI with TA and subsequently with 3-amino-1-propanol. Excess amount of 3-amino-1-propanol was used to ensure the complete aminolysis of PSI.50 The chemical structure of PASP-g-TA/AP was confirmed by 1H NMR in D2O (Fig. 1: δ (ppm) 1.69 (2H, C–CH2–C), 3.23 (4H, C–CH2–N), 3.57 (2H, C–CH2–O)). The peak at 4.65 ppm is corresponded to the methine in the repeating units of the polyaspartamide backbone. The broad peak in the 2.5–3.0 ppm was due to the overlap of phenol group and methylene in the repeating units of the polyaspartamide backbone. The appearance of peaks between 6.6 and 7.2 ppm was attributed to the TA moieties introduced. The disappearance of the peak at 5.1–5.4 ppm indicated the complete aminolysis of PSI. The degree of substitution (DS) of PASP-g-TA/AP determined by comparing the integrals of signals at 6.6–7.2 ppm and 1.69 ppm indicates that the DS consistently matches with feed ratio of TA, which means the DS can be well controlled by the feed ratio of TA.
 | ||
| Scheme 2 Synthetic route of PASP-g-TA/AP. | ||
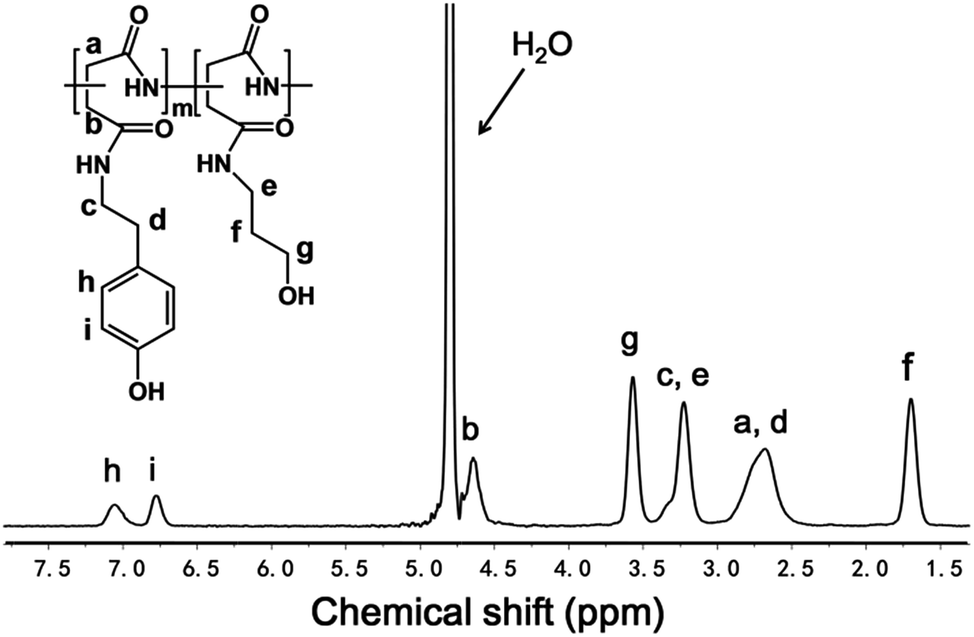 | ||
| Fig. 1 1H NMR spectrum of the PASP-g-TA/AP. | ||
The molecular weights and the DS of TA were summarized in Table 1. As is shown, after the aminolysis with TA and 3-amino-1-propanol, the Mw and Mn of the polymer decreased to 32.9 × 103 and 25 × 103, which meant the chain scission of polymer in some degree. PSI is susceptive to alkaline compound and stronger alkaline tyramine will result in degradation of the polyamide chain in certain degree.55
| Polymer | Mn (×103) | Mw (×103) | Mw/Mn | DS (TA) (%) |
|---|---|---|---|---|
| PSI | 28.5 | 36.8 | 1.3 | — |
| PASP-g-TA/AP | 25.0 | 32.9 | 1.3 | 19 |
3.2 Hydrogel formation and gelation time determination
As appropriate gelation time is required for injectable hydrogels in biomedical applications, the gelation time of PASP-g-TA/AP hydrogel was investigated. The different compositions of hydrogel samples were summarized in ESI Table S1.† Due to the high gelation rate of some samples, the vial-tilting method instead of rheometry was used to determine the gelation time. As shown in Fig. 2a, the gelation time decreased with increasing HRP concentration and decreasing H2O2 concentration when the polymer concentration was fixed at 4.1%. This result should be ascribed to the fact that HRP acted as a catalyst in the reaction. Thus the increased HRP concentration accelerated the generation of the phenoxy radical by H2O2 in the coupling reaction more effectively, resulting in faster gelation.56 On the contrary, excess H2O2 further combined with HRP to form an inactive compound and inhibited the activity of HRP, leading to a longer gelation time.41 The relationship between the gelation time and the polymer concentration was also studied as shown in Fig. 2b. The gelation time decreased at higher polymer concentration. This phenomenon should be attributed to the more crosslinking sites offered by the increased content of phenol group in the precursors solution.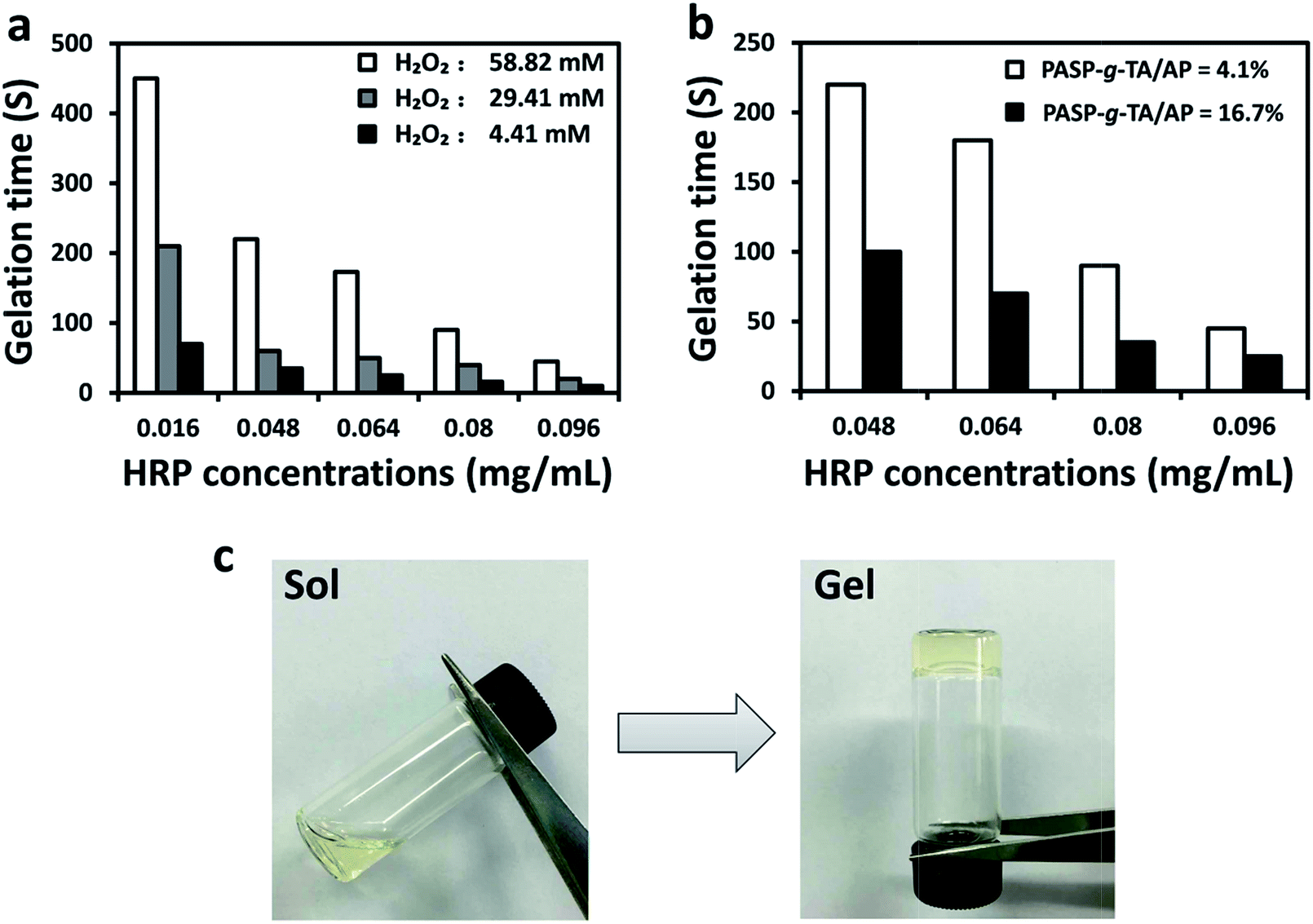 | ||
| Fig. 2 (a) Plot of gelation time of PASP-g-TA/AP hydrogels on different H2O2 concentrations of 58.82, 29.41 and 4.41 mM and different HRP concentrations ranged from 0.016 to 0.096 mg mL−1, [polymer] = 4.1%. (b) Plot of gelation time on different polymer concentrations of 16.7 and 4.1%, [H2O2] = 58.82 mM. (c) Photographs of precursors solution before and after gelation, [polymer] = 4.1%, [HRP] = 0.016 mg mL−1, [H2O2] = 4.41 mM. | ||
3.3 Morphology of the hydrogels
The morphology of the PASP-g-TA/AP hydrogels prepared with different compositions was observed by SEM (Fig. 3). Three-dimensional (3D) porous structure was obvious in Fig. 3a–c and the porous structure was closely related to the concentrations of the polymer, HRP and H2O2. It is known that crosslink density often exerts influence on the pore volume and pore size of porous material. The pore wall of the hydrogel containing 4.1% of polymer, 29.41 mM of H2O2 and 0.064 mg mL−1 of HRP is the thinnest and the porous size is the largest compared to the others as shown in Fig. 3a. When the concentrations of polymer and H2O2 increased from 4.1 to 16.7% and 29.41 to 58.82 mM respectively at a fix HRP concentration of 0.064 mg mL−1, the pore wall of the hydrogel became much thicker and tougher (Fig. 3b), the pore size was also obviously smaller than that in Fig. 3a, indicating the increased crosslink density in the hydrogel fabricated at higher concentrations of polymer and H2O2. Seen from Fig. 3c, however, when the H2O2 concentration further increased from 58.82 to 141.11 mM and the concentrations of polymer and HRP were kept the same as those in Fig. 3b, the pore size seems larger and not uniform, which implied that the excess amount of H2O2 was detrimental to the formation of tight and uniform inner structure of hydrogel. By further comparing Fig. 3d to c, with the HRP concentration decreasing from 0.064 to 0.016 mg mL−1, the hydrogel processed very week network and not typical macroporous structure. These results demonstrated that crosslinking density was affected by the concentrations of polymer, HRP and H2O2, higher concentrations of polymer, HRP and H2O2 contributed to the increased crosslink degree, thus more stable and tough porous structure was formed in the hydrogel. Nevertheless, the excess H2O2 damaged the uniform and tightness of hydrogel inner structure because of its inhibitory effect on HRP, which reduced the crosslink density. In general, pore structure of a hydrogel can influence chemical exchange between its surface and interior, subsequently the release behavior of drugs and the survival, migration, proliferation and differentiation of cells.57–59 In this study, the inner pore-size of the hydrogel could be easily regulated and controlled by adjusting the concentrations of PASP-g-TA/AP, HRP and H2O2, which makes great sense to the further application.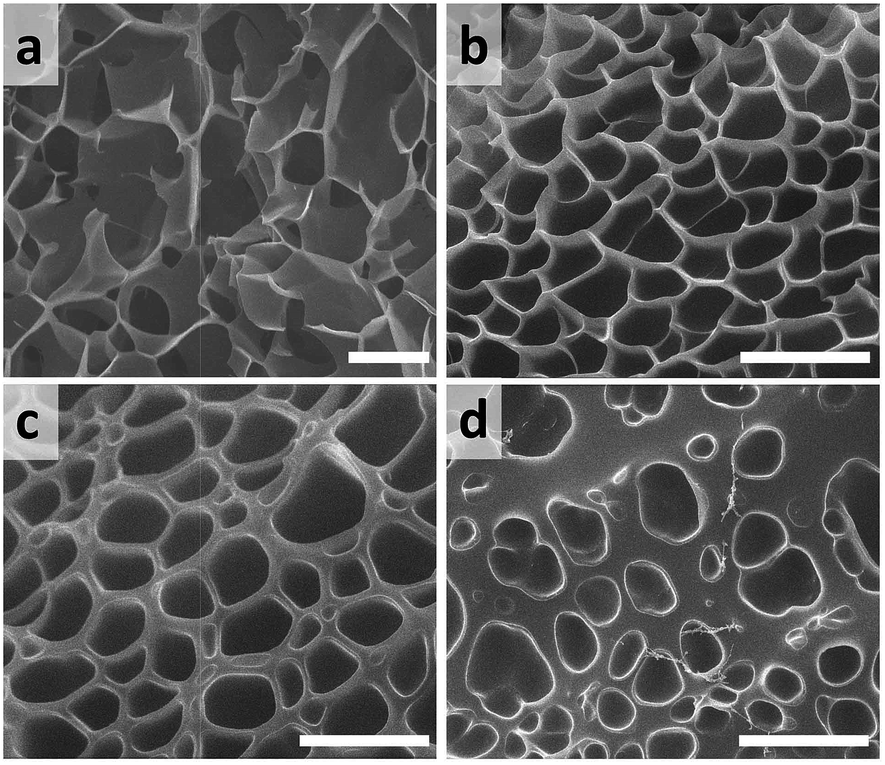 | ||
| Fig. 3 SEM images of hydrogel samples (a) Gel 1-2-4, (b) Gel 2-2-2, (c) Gel 2-4-3, (d) Gel 2-4-1. Scale bar: 10 μm. The formulas of hydrogels were presented in ESI Table S1.† | ||
3.4 Rheological experiments
The rheological properties of PASP-g-TA/AP hydrogels were investigated in a time-controlled oscillatory mode at 37 °C. The formation of the hydrogel under the condition of polymer 4.1%, HRP 0.003 mg mL−1 and H2O2 4.41 mM was shown in Fig. 4a. G′ and G′′ represented the storage modulus and loss modulus of the hydrogel, respectively. It was found that the crossover of G′ and G′′ was observed immediately with a 45° phase angel. Since the crossover point was defined as the gelation point,60 the result suggested that a fast crosslinking process occurred as soon as the mixture solution was loaded on the plate of rheometer. As time progressed, G′ increased until reaching a plateau after 7 minutes and the phase angle was close to zero, which demonstrated the complete formation of the hydrogel.61 The impacts of the concentrations of polymer, HRP and H2O2 on G′ were further investigated. As shown in Fig. 4b, the hydrogel fabricated at 29.41 mM of H2O2 showed the highest G′ when the concentrations of polymer and HRP were fixed at 4.1% and 0.064 mg mL−1 respectively. This may be ascribed to the impact of H2O2 concentration on the crosslink density in enzyme-mediated reaction. In a certain range, the higher H2O2 concentration of 29.41 mM increased the crosslinking density through generating more phenoxy radicals in the coupling reaction, but the excess amount of H2O2 (88.23 mM) inhibited the activity of HRP and reduced the crosslink density.50 The G′ of the hydrogel was positively correlated with HRP concentration as shown in Fig. 4c. With the HRP concentration increasing from 0.016 to 0.064 mg mL−1 at 4.1% of polymer and 29.41 mM of H2O2, G′ varied from 174 to 988 Pa. This phenomenon may be explained by that the higher HRP concentration catalyzed more H2O2 and improved the crosslink reaction more effectively and completely, thus the G′ of the hydrogel increased. The same correlation between G′ and polymer concentration at fixed concentrations of HRP and H2O2 was observed in Fig. 4d. The G′ of the hydrogel significantly strengthened from 497 to 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 873 Pa as the polymer concentration ranged from 4.1 to 16.7%. This should be owed to that the existence of high concentration of phenol groups led to increased crosslink density. Therefore, the viscoelastic property of PASP-g-TA/AP hydrogel was controlled by the concentrations of polymer, HRP and H2O2, among which the polymer concentration was the dominant factor.
873 Pa as the polymer concentration ranged from 4.1 to 16.7%. This should be owed to that the existence of high concentration of phenol groups led to increased crosslink density. Therefore, the viscoelastic property of PASP-g-TA/AP hydrogel was controlled by the concentrations of polymer, HRP and H2O2, among which the polymer concentration was the dominant factor.
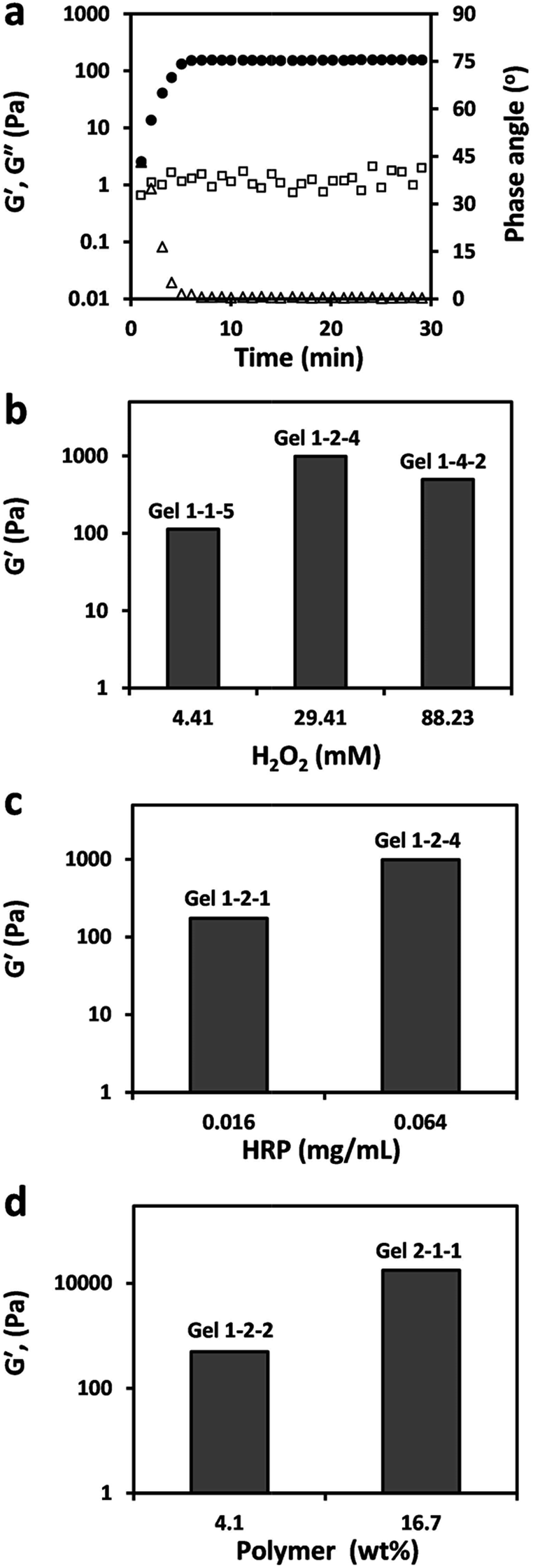 | ||
| Fig. 4 (a) Typical evolution of the storage modulus G′ (●), loss modulus G′′ (□) and phase angle δ (△) of Gel 1-1-1 as a function of time. Storage modulus G′ of hydrogel as a function of (b) [H2O2]; (c) [HRP]; (d) [polymer]. The formulas of hydrogels were presented in ESI Table S1.† | ||
3.5 Swelling ratio
The swelling behaviors of the PASP-g-TA/AP hydrogel were studied by measuring the weight of the gels in phosphate buffer at predetermined time intervals. As shown in Fig. 5a, all the hydrogels reached their equilibrium swelling state in 25 h. The equilibrium swelling ratio ranged from 5 to 42 as polymer concentration changed from 16.7 to 4.1% when the concentrations of HRP and H2O2 were fixed at 0.064 mg mL−1 and 88.23 mM respectively (Fig. 5b). Besides, with the HRP concentration increasing from 0.032 to 0.096 mg mL−1 when the hydrogels were synthesized at the same concentrations of polymer and H2O2, the equilibrium swelling ratio decreased from 46 to 31 (Fig. 5c). These observations may be attributed to the fact that the higher crosslinking density brings the lower equilibrium swelling ratio. Hydrogels formed with the increased content of polymer and HRP had higher crosslinking density, thus the equilibrium swelling ratio reduced. Fig. 5d showed the equilibrium swelling ratio of the hydrogels formed with increased H2O2 concentration while the concentrations of polymer and HRP were kept the same. The sample which H2O2 concentration is 29.41 mM had the lowest swelling ratio because of its highest crosslink density. These observations are in agreement with the results from rheological study.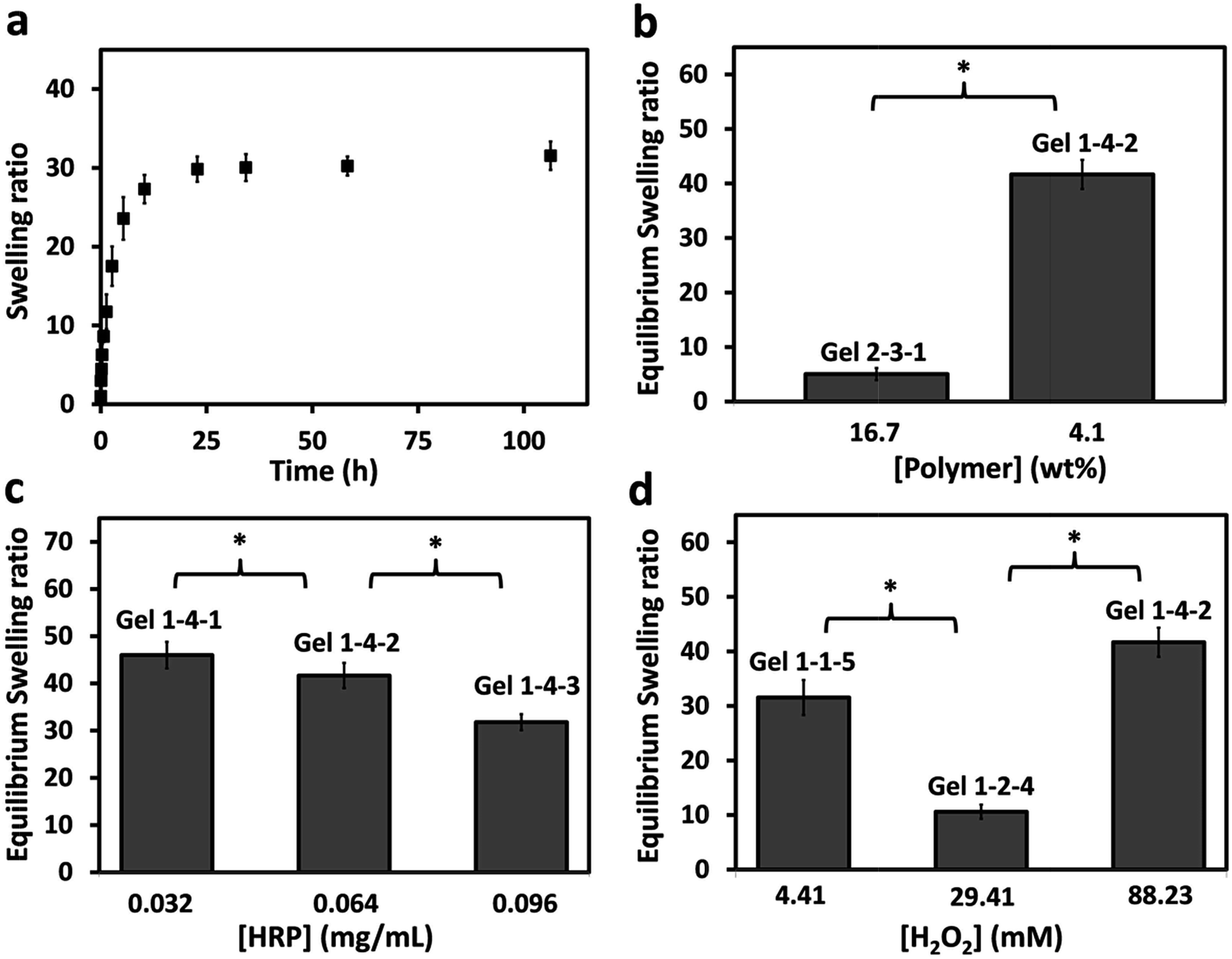 | ||
| Fig. 5 (a) Swelling kinetics of hydrogel sample Gel 1-1-5 in PBS at 37 °C. Equilibrium swelling ratios of hydrogel as a function of (b) [polymer]; (c) [HRP]; (d) [H2O2]. The formulas of hydrogels were presented in ESI Table S1.† (*p < 0.05). | ||
3.6 In vitro drug release
Rh B was chosen as a water-soluble model drug loaded into the hydrogels to estimate the drug release behavior in vitro of PASP-g-TA/AP hydrogels. The influences of the polymer, HRP and H2O2 concentrations on the drug release rate were assessed (see Fig. 6). As shown in Fig. 6a, all the samples containing 4.1% of polymer exhibited an initial rapid release of drugs during first 20 h, and the cumulative release of Rh B reached steady-state after 60 h. When the H2O2 concentration was fixed at 4.41 mM and the HRP concentration increased from 0.016 to 0.064 mg mL−1 (Gel 1-1-2 and Gel 1-1-5), the two curves almost overlapped. When the HRP concentration was fixed at 0.064 mg mL−1 (Gel 1-1-5, Gel 1-5-1 and Gel 1-2-4), the hydrogel formed at 29.41 mM of H2O2 showed the slowest release rate and that prepared at 141.11 mM of H2O2 exhibited the fastest release rate. However, quadrupling the polymer concentration from 4.1 to 16.7% at fixed concentrations of HRP and H2O2 as shown in Fig. 6b, an almost zero-order release profile was observed instead of the simple diffusion-based release behavior during the first 17 days and Rh B was released sustainedly for 27 days. The results in this study indicated that the polymer concentration had the most significant effect on Rh B release rate and the hydrogel formed at 16.7% of polymer displayed good controlled release properties for the model drugs. These results could be explained from the hydrogen bonds formed between the –COOH from Rh B and the –OH from PASP-g-TA/AP in the network. When Rh B released from the network of PASP-g-TA/AP hydrogel, the Rh B needed to overcome stronger interaction with the hydrogel containing more –OH due to the increased polymer concentration, making the hydrogel fabricated at 16.7% of polymer concentration showed much slower release rate than those fabricated at 4.1% of polymer concentration. Besides, crosslinking density of hydrogel is recognized as one of the most important parameter in affecting the drugs delivery behavior and higher crosslinking density usually gives a slower release rate. Fixing the polymer concentration at 4.1%, the sample with higher crosslink density exhibited lower swelling ratio, which means there are higher polymer content and therefore more –OH groups in unit volume of release medium. With the drug-loaded hydrogel swelling in PBS, Rh B diffused to the outside solution more hardly from the hydrogel network with higher crosslinking density due to the hydrogen bond interaction. As a result, the sample fabricated at 0.064 mg mL−1 of HRP and 29.41 mM of H2O2 showed slowest release rate owing to its highest crosslinking density. However, when the concentrations of polymer and H2O2 were kept at 4.1% and 4.41 mM respectively, the drug release behavior of samples with different HRP concentrations were almost the same. This phenomenon should attribute to the relatively small impact of HRP concentration on crosslinking density and swelling ratio, which make the influence of HRP concentration on release rate was not obvious.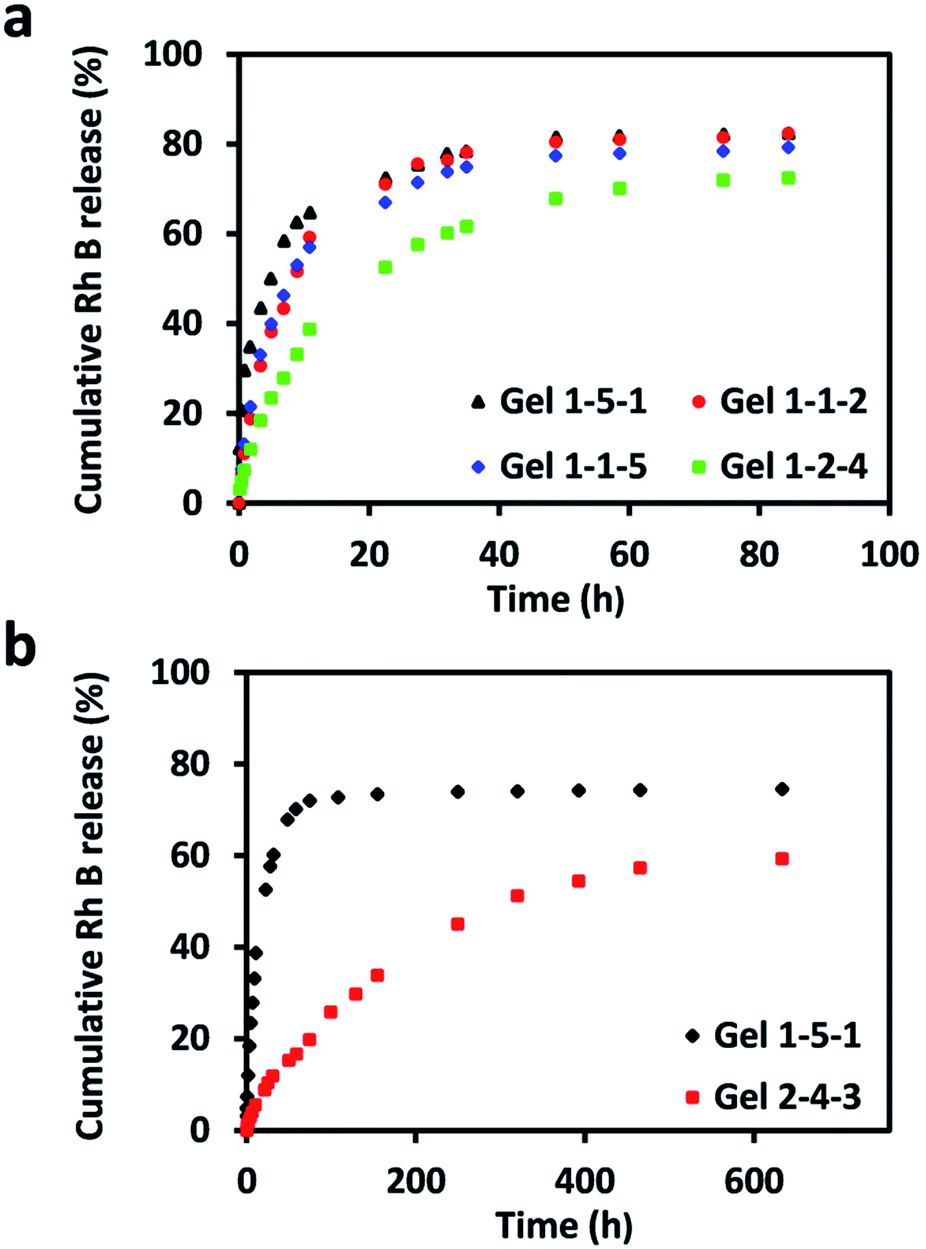 | ||
| Fig. 6 In vitro release percentage of Rh B (analyzed by UV spectrum) from different hydrogels. Hydrogels with (a) [polymer] = 4.1%; (b) [H2O2] = 141.11 mM, [HRP] = 0.064 mg mL−1. The formulas of hydrogels were presented in ESI Table S1.† | ||
3.7 In vitro cytotoxicity
The cytotoxicity study of the hydrogel was performed using MTT assay. The results in Fig. 7a demonstrated good cytocompatibility of PASP-g-TA/AP hydrogel disks as the viability of the COS-7 cells were all over 100% at the hydrogel concentrations ranging from 16 to 36 g L−1. This phenomenon could be attributed to the full contact between the submerged hydrogel disks and cells, which made the cells under the disks aggregate and grow towards the hydrogel while the cells in control group suffered from area restriction of TCPS after culturing for 48 h. Besides, Fig. 7b displayed the cytotoxicity of the extract of PASP-g-TA/AP hydrogels. The cell viability had reached to about 90% at the concentrations of hydrogel extract solution ranging from 20 to 500 mL L−1, which indicated low cytotoxicity of the extract of PASP-g-TA/AP hydrogel.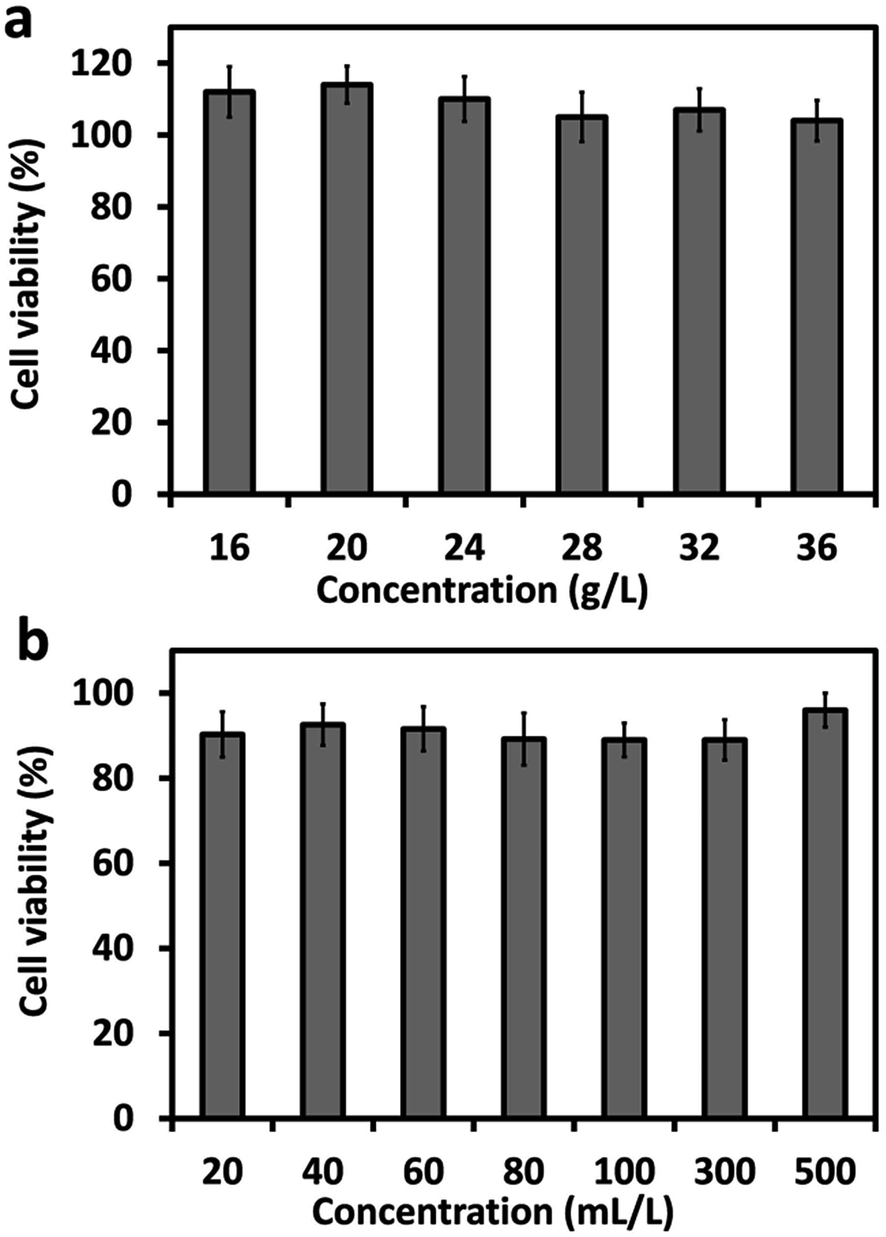 | ||
| Fig. 7 In vitro cytotoxicity of (a) PASP-g-TA/AP hydrogel disks and (b) the extract solution of PASP-g-TA/AP hydrogel to COS-7 cells. | ||
3.8 3D cell culture within PASP-g-TA/AP hydrogels
3D culture study of COS-7 cells inside the hydrogels was used to evaluate the cell viability and proliferation within the hydrogels. COS-7 cells were incorporated inside the PASP-g-TA/AP hydrogels during the gel preparation and cultured up to 7 days. AO/EB was used to distinguish the live and dead cells, which stained the live cells with green and dead ones with red. The cell viability and proliferation were observed by CLSM on days 1, 3 and 6. As shown in Fig. 8a, most of the cells presented green, which indicated a high viability of the cells cultured within the hydrogels. In addition, the cells aggregated over time, which could be explained by the weak interaction between COS-7 cells and the hydrogel due to the high hydrophilicity of PASP-g-TA/AP gels.62–64 Cell proliferation cultured within the hydrogel compared with that on TCPS was further evaluated by CCK-8 method on days 1, 3, 5 and 7. As shown in Fig. 8b, the COS-7 cells cultured in the hydrogels remained viable and exhibited slow growth rate at first and sustainable proliferation during the whole culture time of 7 days. On the contrary, the cells cultured on TCPS grew fast during the first three days but barely increased from day 3 to day 7. This result may be explained by the cell polarization forced by culturing on 2D surface. The cells cultured on TCPS experienced unnatural interactions with the environment and neighboring cells,65 making them fully contact with the nutrient of culture medium, whereas the cells cultured in hydrogel tended to aggregate, preventing the exchange of culture medium to a certain degree. As a result, the cells cultured on TCPS grew much faster than that cultured within the hydrogel during the first three days. However, when cells continued to grow, the area reduction of the TCPS hindered the contact between cells and the culture medium that largely reduced the proliferation rate. On the other hand, hydrogel provided 3D expanded space for cells to grow without restriction on effective transportation of culture medium, carbon dioxide and oxygen. Thus, the cells cultured within PASP-g-TA/AP hydrogel displayed sustainable growth. In the study of 3D culture and the in vitro cytotoxicity, the PASP-g-TA/AP hydrogel has revealed good compatibility for the potential use in biomedical applications.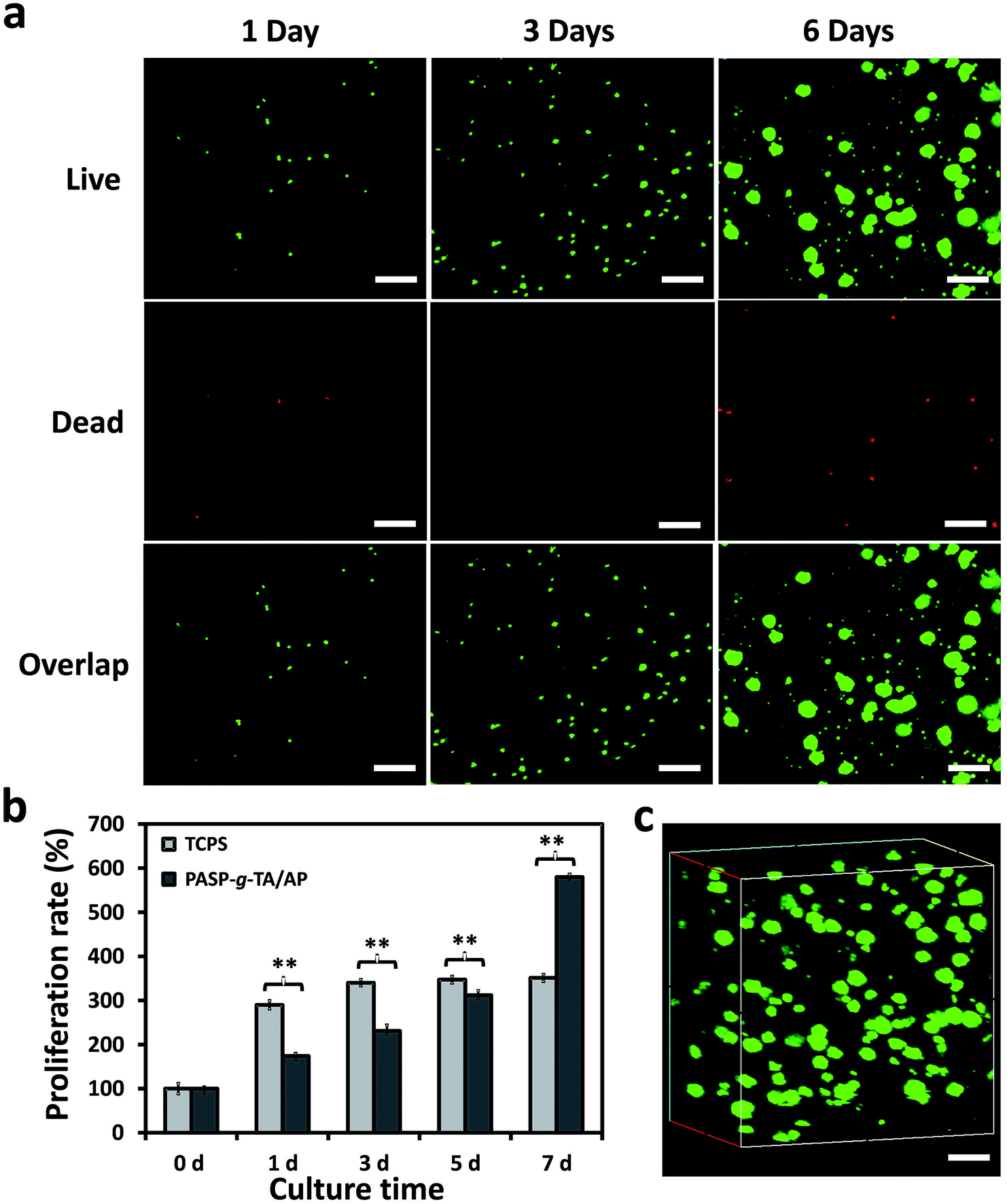 | ||
| Fig. 8 (a) CLSM images of COS-7 cells cultured in PASP-g-TA/AP hydrogel for 1, 3 and 6 days. The cells were double stained with AO/EB. (b) Proliferation rate of COS-7 cells at different culture time with cells cultured on normal 96-well TCPS and in PASP-g-TA/AP hydrogel. Data was analyzed by the CCK-8 method (n = 3) (**p < 0.01). Cell seeding density: 3.3 × 105 mL−1. (c) 3D view of CLSM image of COS-7 cells cultured in the hydrogel for 6 days. Cells were double stained with AO/EB. Scale bar: 200 μm. | ||
3.9 In vivo gel formation and biocompatibility
To investigate the in vivo gel formation of PASP-g-TA/AP hydrogel, 210 μL of mixed solution in PBS containing polymer (4.1%), H2O2 (4.41 mM) and HRP (0.016 mg mL−1) was injected into the subcutaneous layer of mice. 30 minutes later, a mouse was sacrificed and the hydrogel was found in the subcutaneous layer of the mouse (Fig. 9a1), which indicated that PASP-g-TA/AP solution could be easily injected and formed a hydrogel rapidly in vivo in the presence of HRP and H2O2. The hydrogels were maintained and observed for 30 days (Fig. 9a2). For the in vivo biocompatibility study of PASP-g-TA/AP hydrogels, mice were sacrificed at different time intervals and the tissues containing the gels were removed and stained with H&E to examine the histological response. There can be found more glands than those in normal tissue without gel in the corium layer (Fig. 9b1) and increased number of inflammatory cells surrounding the gel (Fig. 9b2) in the first 2 days after injection, which indicated the inflammatory reaction in the initial stage.66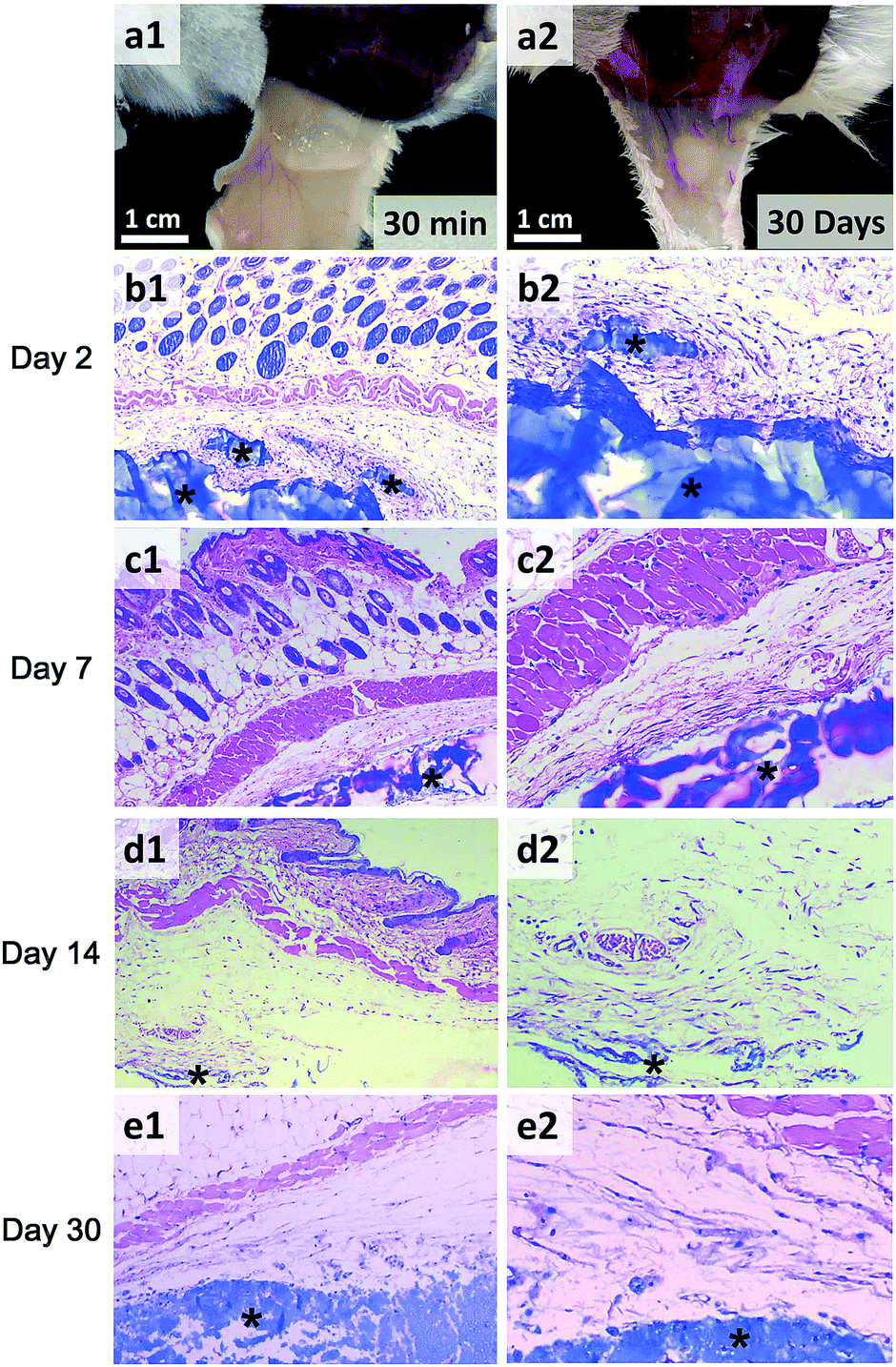 | ||
| Fig. 9 In vivo PASP-g-TA/AP hydrogel formation and tissue biocompatibility. The image of hydrogel was taken 30 minutes (a1) and 30 days (a2) after subcutaneous injection. The images of H&E staining of the surrounding tissues 2 days (b1, b2), 7 days (c1, c2), 14 days (d1, d2) and 30 days (e1, e2) post-injection. b1, c1, d1 and e1: 40×; b2, c2, d2 and e2: 100×. Hydrogels are marked with asterisks (*). | ||
The number of inflammatory cells and glands decreased after 7 days (Fig. 9c1 and c2) and almost reduced to the normal state (Fig. 9d1 and d2) after 14 days. These results implied that acute inflammation in the subcutaneous tissues would regress spontaneously in 2 weeks. On the thirtieth day after subcutaneous injection, notable degradation of the hydrogel caused by mammalian proteolytic enzymes41,67 was observed as shown in Fig. 9a2, e1 and e2. Besides, there was no obvious tissue necrosis, edema, hyperemia, hemorrhaging and muscle damage observed during the experimental period. Thus, the results suggested that the PASP-g-TA/AP hydrogel may have the potential in 3D cell culture and tissue engineering.
4 Conclusions
An injectable and biodegradable hydrogel was successfully prepared by enzymatic crosslinking of TA modified PSI. The gelation time and the viscoelastic property of PASP-g-TA/AP hydrogel were depended on the concentrations of polymer, HRP and H2O2. The physicochemical properties including hydrogel morphology and swelling ratio could be easily adjusted through varying the concentrations of polymer, HRP and H2O2. Especially in the study of drug release behavior, the hydrophilic small molecule drugs could be loaded into the hydrogel by simply mixing, and the hydrogel displayed good controlled release properties without bursting release. In vitro cytotoxicity test showed that the PASP-g-TA/AP hydrogel had low cytotoxicity to COS-7 cells. In 3D culture study, the COS-7 cells cultured within the hydrogel presented high viability and showed a sustainable proliferation during the culture period of 7 days. Besides, the hydrogels formed in the subcutaneous layer of mouse showed acceptable biocompatibility and biodegradability for up to 30 days. In conclusion, the injectable and biodegradable PASP-g-TA/AP hydrogels formed by enzymatically crosslinking offered great potential as a promising biomaterial for localized and long-term sustained delivery of hydrophilic drugs and 3D scaffolds for tissue engineering.Acknowledgements
Financial support from National Natural Science Foundation of China (51533006) is gratefully acknowledged.References
- H. T. Ta, C. R. Dass, I. Larson, P. F. M. Choong and D. E. Dunstan, Biomaterials, 2009, 30, 4815–4823 CrossRef CAS PubMed.
- D. Puppi, C. Migone, L. Grassi, A. Pirosa, G. Maisetta, G. Batoni and F. Chiellini, Polym. Int., 2016, 65, 631–640 CrossRef CAS.
- M.-G. Kim, Y. Shon, W. Miao, J. Lee and Y.-K. Oh, Carbon, 2016, 105, 14–22 CrossRef CAS.
- H. Hamidian and T. Tavakoli, Carbohydr. Polym., 2016, 144, 140–148 CrossRef CAS PubMed.
- M. Dehghan-Niri, M. Tavakol, E. Vasheghani-Farahani and F. Ganji, J. Biomater. Appl., 2015, 29, 1343–1350 CrossRef CAS PubMed.
- D. Ma, G. An, M. Liang, Y. Liu, B. Zhang and Y. Wang, Mater. Sci. Eng., C, 2016, 65, 221–231 CrossRef CAS PubMed.
- T. Billiet, M. Vandenhaute, J. Schelfhout, S. Van Vlierberghe and P. Dubruel, Biomaterials, 2012, 33, 6020–6041 CrossRef CAS PubMed.
- P. Baei, S. Jalili-Firoozinezhad, S. Rajabi-Zeleti, M. Tafazzoli-Shadpour, H. Baharvand and N. Aghdami, Mater. Sci. Eng., C, 2016, 63, 131–141 CrossRef CAS PubMed.
- K. M. Park, Y. Lee, J. Y. Son, D. H. Oh, J. S. Lee and K. D. Park, Biomacromolecules, 2012, 13, 604–611 CrossRef CAS PubMed.
- H. J. van der Linden, S. Herber, W. Olthuis and P. Bergveld, Analyst, 2003, 128, 325–331 RSC.
- K. L. Wang, J. H. Burban and E. L. Cussler, Adv. Polym. Sci., 1993, 110, 67–79 CrossRef CAS.
- D. Jaikumar, K. M. Sajesh, S. Soumya, T. R. Nimal, K. P. Chennazhi, S. V. Nair and R. Jayakumar, Int. J. Biol. Macromol., 2015, 74, 318–326 CrossRef CAS PubMed.
- A. Navaei, D. Truong, J. Heffernan, J. Cutts, D. Brafman and R. W. Sirianni, et al., Acta Biomater., 2016, 32, 10–23 CrossRef CAS PubMed.
- H. Liu, J. Liu, C. Qi, Y. Fang, L. Zhang and R. Zhuo, et al., Acta Biomater., 2016, 35, 228–237 CrossRef CAS PubMed.
- J. Thiele, Y. Ma, S. M. C. Bruekers, S. Ma and W. T. S. Huck, Adv. Mater., 2014, 26, 125–148 CrossRef CAS PubMed.
- H. Liu, Q. Yang, L. Zhang, R. Zhuo and X. Jiang, Carbohydr. Polym., 2016, 137, 600–607 CrossRef CAS PubMed.
- X. Wang, H. Hu, W. Wang, K. I. Lee, C. Gao, L. He, Y. Wang, C. Lai, B. Fei and J. H. Xin, Colloids Surf., B, 2016, 143, 342–351 CrossRef CAS PubMed.
- Z. Zhang, J. Ni, L. Chen, L. Yu, J. Xu and J. Ding, Biomaterials, 2011, 32, 4725–4736 CrossRef CAS PubMed.
- Y.-L. Chiu, S.-C. Chen, C.-J. Su, C.-W. Hsiao, Y.-M. Chen, H.-L. Chen and H.-W. Sung, Biomaterials, 2009, 30, 4877–4888 CrossRef CAS PubMed.
- O. V. Khutoryanskaya, Z. A. Mayeva, G. A. Mun and V. V. Khutoryanskiy, Biomacromolecules, 2008, 9, 3353–3361 CrossRef CAS PubMed.
- F. Y. Ding, X. Qian, Q. Zhang, H. J. Wu, Y. Y. Liu, L. Xiao, H. B. Deng, Y. M. Du and X. W. Shi, New J. Chem., 2015, 39, 1253–1259 RSC.
- M. Tortora, F. Cavalieri, E. Chiessi and G. Paradossi, Biomacromolecules, 2007, 8, 209–214 CrossRef CAS PubMed.
- G. Xu, X. Wang, C. Deng, X. Teng, E. J. Suuronen, Z. Shen and Z. Zhong, Acta Biomater., 2015, 15, 55–64 CrossRef CAS PubMed.
- D. Mandracchia, G. Pitarresi, F. S. Palumbo, B. Carlisi and G. Giammona, Biomacromolecules, 2004, 5, 1973–1982 CrossRef CAS PubMed.
- F. Y. Ding, S. P. Wu, S. S. Wang, Y. Xiong, Y. Li, B. Li, H. B. Deng, Y. M. Du, L. Xiao and X. W. Shi, Soft Matter, 2015, 11, 3971–3976 RSC.
- J. Liu, K. Wang, J. Luan, Z. Wen, L. Wang, Z. L. Liu, G. Y. Wu and R. X. Zhuo, J. Mater. Chem. B, 2016, 4, 1343–1353 RSC.
- B. Wang, Y. S. Jeon, H. S. Park, Y. J. Kim and J. H. Kim, eXPRESS Polym. Lett., 2015, 9, 799–808 CrossRef CAS.
- A. P. Bapat, D. Roy, J. G. Ray, D. A. Savin and B. S. Sumerlin, J. Am. Chem. Soc., 2011, 133, 19832–19838 CrossRef CAS PubMed.
- S. C. Owen, S. A. Fisher, R. Y. Tam, C. M. Nimmo and M. S. Shoichet, Langmuir, 2013, 29, 7393–7400 CrossRef CAS PubMed.
- C. M. Nimmo, S. C. Owen and M. S. Shoichet, Biomacromolecules, 2011, 12, 824–830 CrossRef CAS PubMed.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592–605 CrossRef CAS PubMed.
- P. V. Chang and C. R. Bertozzi, Chem. Commun., 2012, 48, 8864–8879 RSC.
- R. Kennedy, W. Ul Hassan, A. Tochwin, T. Zhao, Y. Dong, Q. Wang, H. Tai and W. Wang, Polym. Chem., 2014, 5, 1838–1842 RSC.
- L. S. M. Teixeira, J. Feijen, C. A. van Blitterswijk, P. J. Dijkstra and M. Karperien, Biomaterials, 2012, 33, 1281–1290 CrossRef PubMed.
- M. Kurisawa, F. Lee, L. S. Wang and J. E. Chung, J. Mater. Chem., 2010, 20, 5371–5375 RSC.
- R. Jin, L. S. M. Teixeira, P. J. Dijkstra, M. Karperien, C. A. van Blitterswijk, Z. Y. Zhong and J. Feijen, Biomaterials, 2009, 30, 2544–2551 CrossRef CAS PubMed.
- R. Wang, N. Leber, C. Buhl, N. Verdonschot, P. J. Dijkstra and M. Karperien, Polym. Adv. Technol., 2014, 25, 568–574 CrossRef CAS.
- V. Kuzmenko, D. Hagg, G. Toriz and P. Gatenholm, Carbohydr. Polym., 2014, 102, 862–868 CrossRef CAS PubMed.
- M. Kurisawa, J. E. Chung, Y. Y. Yang, S. J. Gao and H. Uyama, Chem. Commun., 2005, 4312–4314, 10.1039/b506989k.
- M. Obst and A. Steinbuchel, Biomacromolecules, 2004, 5, 1166–1176 CrossRef CAS PubMed.
- K. X. Ren, C. L. He, Y. L. Cheng, G. Li and X. S. Chen, Polym. Chem., 2014, 5, 5069–5076 RSC.
- J.-H. Kim, C. M. Son, Y. S. Jeon and W.-S. Choe, J. Polym. Res., 2011, 18, 881–890 CrossRef CAS.
- C. Lu, X. Wang, G. Wu, J. Wang, Y. Wang, H. Gao and J. Ma, J. Biomed. Mater. Res., Part A, 2014, 102, 628–638 CrossRef PubMed.
- B. Gyarmati, E. Krisch and A. Szilágyi, React. Funct. Polym., 2014, 84, 29–36 CrossRef CAS.
- C. Fiorica, F. S. Palumbo, G. Pitarresi, A. Gulino, S. Agnello and G. Giammona, RSC Adv., 2015, 5, 19715–19723 RSC.
- S. Sofia, A. Singh and D. Kaplan, J. Macromol. Sci., Part A: Pure Appl. Chem., 2002, 39, 1151–1181 CrossRef.
- D. Chen, Y. Ping, G. Tang and J. Li, Soft Matter, 2010, 6, 955 RSC.
- G. P. Tang, K. J. Zhu and Q. Q. Chen, J. Appl. Polym. Sci., 2000, 77, 2411–2417 CrossRef CAS.
- C. Li, G.-F. Luo, H.-Y. Wang, J. Zhang, Y.-H. Gong, S.-X. Cheng, R.-X. Zhuo and X.-Z. Zhang, J. Phys. Chem. C, 2011, 115, 17651–17659 CAS.
- S. P. Hsu, I. M. Chu and J. D. Yang, J. Appl. Polym. Sci., 2012, 125, 133–144 CrossRef CAS.
- K. M. Park, Y. M. Shin, Y. K. Joung, H. Shin and K. D. Park, Biomacromolecules, 2010, 11, 706–712 CrossRef CAS PubMed.
- R. Jin, C. Hiemstra, Z. Y. Zhong and J. Feijen, Biomaterials, 2007, 28, 2791–2800 CrossRef CAS PubMed.
- L. Weng, A. Romanov, J. Rooney and W. Chen, Biomaterials, 2008, 29, 3905–3913 CrossRef CAS PubMed.
- J. Trudel and S. P. Massia, Biomaterials, 2002, 23, 3299–3307 CrossRef CAS PubMed.
- K. Seo and D. Kim, Acta Biomater., 2010, 6, 2157–2164 CrossRef CAS PubMed.
- R. Jin, L. S. M. Teixeira, P. J. Dijkstra, C. A. van Blitterswijk, M. Karperien and J. Feijen, Biomaterials, 2010, 31, 3103–3113 CrossRef CAS PubMed.
- F. Lee, J. E. Chung and M. Kurisawa, J. Controlled Release, 2009, 134, 186–193 CrossRef CAS PubMed.
- X. Q. Jia, J. A. Burdick, J. Kobler, R. J. Clifton, J. J. Rosowski, S. M. Zeitels and R. Langer, Macromolecules, 2004, 37, 3239–3248 CrossRef CAS.
- B. B. Mandal, S. Kapoor and S. C. Kundu, Biomaterials, 2009, 30, 2826–2836 CrossRef CAS PubMed.
- H. H. Winter, J. Rheol., 1986, 30, 367 CrossRef CAS.
- F. Lee, J. E. Chung and M. Kurisawa, Soft Matter, 2008, 4, 880–887 RSC.
- J. Lee, M. J. Cuddihy, G. M. Cater and N. A. Kotov, Biomaterials, 2009, 30, 4687–4694 CrossRef CAS PubMed.
- A. Ivascu and M. Kubbies, J. Biomol. Screening, 2006, 11, 922–932 CrossRef CAS PubMed.
- Z. Q. Zhao, J. J. Gu, Y. N. Zhao, Y. Guan, X. X. Zhu and Y. J. Zhang, Biomacromolecules, 2014, 15, 3306–3312 CrossRef CAS PubMed.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS PubMed.
- E. Y. Kang, H. J. Moon, M. K. Joo and B. Jeong, Biomacromolecules, 2012, 13, 1750–1757 CrossRef CAS PubMed.
- C. Li, Adv. Drug Delivery Rev., 2002, 54, 695–713 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra18479k |
| This journal is © The Royal Society of Chemistry 2016 |