
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the thermo- and mechanoresponsive behavior of luminescent cyclophanes†
Yoshimitsu
Sagara
*a,
Christoph
Weder
b and
Nobuyuki
Tamaoki
*a
aResearch Institute for Electronic Science, Hokkaido University, N20, W10, Kita-Ku, Sapporo 001-0020, Japan. E-mail: sagara@es.hokudai.ac.jp; tamaoki@es.hokudai.ac.jp
bAdolphe Merkle Institute, University of Fribourg, Chemin des Verdiers 4, CH-1700 Fribourg, Switzerland
First published on 26th August 2016
Abstract
Many cyclophanes have been investigated in dilute solution, where their internal cavities are accessible for supramolecular interactions. However, their photophysical properties in the solid state remain largely unexplored. We here report a new mechano- and thermoresponsive luminescent cyclophane that is comprised of two 9,10-bis(phenylethynyl)anthracene moieties and features two hexaethylene glycol bridges. The compound was found to exhibit a nematic liquid-crystalline phase at elevated temperature. X-ray diffraction patterns confirm that thermal and mechanical treatments induce changes in the molecular assembly, which are the basis for the observed photoluminescent color variations. The stimuli-responsive behavior of the new compound is quite different from that of a previously reported cyclophane with similar structure but shorter bridges. Thus, merely changing the ring size is an effective tool to tailor the stimuli-responsiveness and the phase behaviour of luminescent cyclophanes.
Introduction
Since the first report on [2.2]paracyclophane in 1949,1 such bridged aromatic compounds have become widely investigated.2 Building on the synthetic pathways pioneered by Cram and Steinberg,3 a large number of cyclophanes have been synthesized over the years, originally on account of a general interest in molecules with unusual shape, high symmetry, and strained character.2,4 Eventually, it was recognized that their cavities render cyclophanes useful as supramolecular hosts, which have the ability to include guest molecules or ions.2,4b,c,5,6 This propensity was widely exploited by using cyclophanes as part of interlocked molecules such as catenanes and rotaxanes.6f,7 In cases where the cyclophane comprises a fluorophore, the photoluminescence properties often change upon formation of inclusion complexes.6e,8–12 This behavior renders cyclophanes useful as fluorescent sensors, e.g., for the detection of biologically important ions and molecules.6e,8–11 As a result, most studies on luminescent cyclophanes have focused on their investigation in solution, while their solid-state properties have remained little examined.13,14Many compounds have been reported to display thermo- and mechanoresponsive luminescence characteristics, and which are possibly useful as sensing materials, in storage devices, and for security applications.15–20 In many cases, a stimuli-induced re-arrangement of the molecular assembly is the cause for the observed changes of the photoluminescence, because the photophysical properties often strongly depend on the molecular arrangement in the solid state.15 Thus, a reliable design strategy for thermo- and/or mechanoresponsive luminescent molecular materials is to create molecules that can assemble in several thermodynamically (meta)stable states.15a Many research groups have shown that this is generally possible by balancing the competition of different intermolecular interactions or using specific molecular structures that induce aggregation-induced emission properties.15a
With the objective to demonstrate that the integration of luminescent motifs into cyclic structures is another general approach to create stimuli-responsive luminescent compounds, we here report on the stimuli-responsive behaviour of the luminescent cyclophane 1 (Fig. 1). The new cyclophane is comprised of two 9,10-bis(phenylethynyl)anthracene moieties, which are connected by two hexaethylene glycol bridges. Unlike the linear reference compound 2, cyclophane 1 displays mechano- and thermoresponsive luminescence in the solid state. This finding follows our recent discovery of the thermo- and mechanoresponsive behavior of cyclophane 3, which features the same luminophores as 1, but shorter bridges.14 Unfortunately, 3 displays a very low solubility in common organic solvents. We show here that the modification of the bridges between the 9,10-bis(phenylethynyl)anthracene moieties leads to an increase of the solubility, but more importantly, a completely different stimuli-responsive phase transition and photoluminescence color changing behavior were observed. Furthermore, compound 1 was found to exhibit a nematic liquid-crystalline (LC) phase.
 | ||
| Fig. 1 Molecular structures of cyclophane 1, the linear reference compound 2, and previously reported cyclophane 3. | ||
Results and discussion
Hexaethylene glycol linkers were selected to bridge two 9,10-bis(phenylethynyl)anthracene moieties to form compound 1. The flexible linkers were extended relative to those used in 3 and expected to enhance the solubility in common solvents and to decrease the phase transition temperatures. The linear compound 2 featuring the same luminophore was also synthesized for reference purposes. Compounds 1 and 2 were characterized by 1H NMR, 13C NMR, MALDI-TOF-MS spectra, and elemental analysis (see ESI†). We found that compound 1 dissolves well in chloroform, dichloromethane, tetrahydrofuran, and N,N-dimethylformamide, whereas hot chloroform was needed to dissolve the previously reported cyclophane 3.14Upon precipitating 1 from a concentrated chloroform solution into hexane, a yellow powder showing yellow photoluminescence was obtained (hereafter referred to as the Y-form). The Y-form can be converted to a yellowish-green emissive form (YG-form) through annealing at 150 °C for 10 min (Fig. 2). In the case of 3, thermal treatment results in a conversion from a yellow emissive crystal to a reddish-orange emissive crystal.14 Thus, the direction of the thermally induced photoluminescence spectral shift of 1 is opposite to that of 3. Upon mechanically grinding the Y- or YG-form of 1, a yellow emissive amorphous form (Am-form) was obtained. When the Am-form was annealed at 150 °C for 10 min, a mixture was obtained that showed both yellow and green emission. Though the Y-form never recovers after thermal or mechanical treatment in the solid state, reprecipitation regenerates the Y-form. In sharp contrast to cyclophane 1, the reference compound 2 does not display similar changes of its photoluminescence color in response to thermal or mechanical stimuli in the solid state. Motivated by these differences, we investigated the relationship between the photophysical properties and the molecular assembled structures of cyclophane 1 in more detail.
 | ||
| Fig. 2 Images presenting the photoluminescence color changes of cyclophane 1 in response to thermal and mechanical stimuli. Arrows indicate possible conversions by the indicated treatments. Images were taken on quartz substrates under irradiation with UV light (λex = 365 nm) at room temperature. | ||
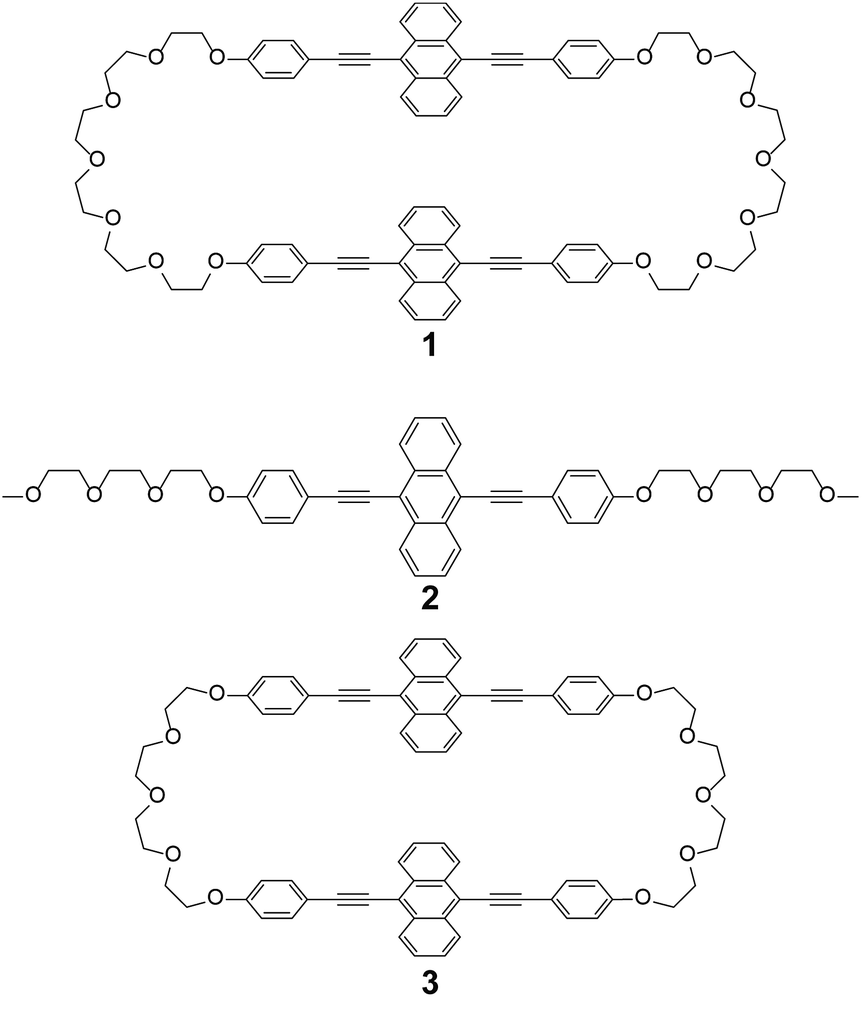
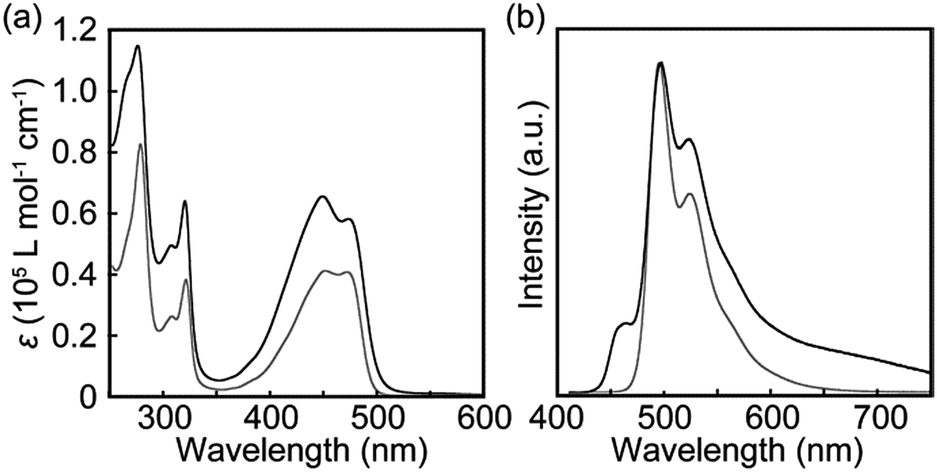
The photophysical properties of cyclophane 1 and the linear reference compound 2 were first probed in dilute chloroform solutions (c = 1 × 10−5 M). The absorption and emission spectra of 2 show the typical spectral features of the 9,10-bis(phenylethynyl)anthracene motif (Fig. 3, grey line). The absorption band displays two peaks at 450 and 471 nm with similar molar absorption coefficient (ε = 4.1 × 104 L mol−1 cm−1, 4.0 × 104 L mol−1 cm−1, respectively) (Fig. 3a). A vibronic structure was observed in the emission band with two peaks at 493 and 522 nm and a shoulder around at 560 nm (Fig. 3b). Fig. 4a shows the result of an emission lifetime measurement conducted for a dilute chloroform solution of 2 (ca. 1 × 10−6 M). The decay profile is well fitted by a single exponential decay function with a lifetime of 3.1 ns. These results match literature data of a similar linear compound well21 and indicate that compound 2 is indeed well-individualized in the chloroform solutions measured.
 | ||
| Fig. 3 (a) Absorption and (b) photoluminescence spectra of chloroform solutions of cyclophane 1 (c = 1 × 10−5 M; black line) and the linear reference compound 2 (c = 1 × 10−5 M; grey line). All spectra were recorded at room temperature. Photoluminescence spectra were recorded with excitation at 400 nm and were normalized to the intensity of the monomer peak. | ||
 | ||
| Fig. 4 Emission decay profiles of chloroform solutions of (a) the linear reference compound 2 collected at 500 nm and (b) cyclophane 1 collected at 500 nm (black line) and 650 nm (grey line). All profiles were measured at room temperature with λex = 405 nm and at concentrations of ca. 1 × 10−6 M. | ||
The chloroform solution of 1 displays an absorption spectrum which is slightly different from that of compound 2 (Fig. 3a). Two peaks are observed at 449 and 474 nm with extinction coefficients, ε, of 6.5 × 104 L mol−1 cm−1 and 5.8 × 104 L mol−1 cm−1. The fact that they are different and not twice as large as those of compound 2 is indicative of electronic ground-state interactions between the two chromophores in cyclophane 1. Similar changes of the absorption spectral shapes have been observed for other cyclophanes that contain two perylene moieties and in which the chromophores form an H-type geometry.22
The emission spectrum of the chloroform solution of 1 is also unambiguously different from that of 2, as shown in Fig. 3b. Besides the monomer emission peak at 493 nm, a new band with maximum at 464 nm is observed, which is ascribed to exciton coupling with H-type geometry of the two emission cores; this interpretation is supported by the fact that the molar extinction coefficient at 471 nm is larger than that at 450 nm (Fig. 3a). Moreover, such an emission peak at shorter wavelength region was also observed for compound 3.14 In ideal H-aggregates, radiative relaxation from the higher energy level resulting from splitting of the lowest unoccupied molecular orbital of monomer is forbidden.23 However, twisted arrangements of the luminophores allow direct radiative transition from the forbidden state. Because the absorption spectral feature does not change and the emission peak at 464 nm remains unchanged in the concentration range from 1 × 10−5 M to 1 × 10−6 M (Fig. S1†), the exciton coupling must arise from an intramolecular H-type geometry and not intermolecular interactions.
Another characteristic of the emission spectrum of the chloroform solution of 1 is the non-negligible emission intensity between 600 and 750 nm (Fig. 3b), which is ascribed to the formation of excimers.24 Indeed, a longer emission lifetime of 8.6 ns was observed when the emission decay profile was monitored at 650 nm (Fig. 4b, grey line), whereas much shorter lifetimes (0.2, 0.6, and 2.2 ns) were obtained when the decay was monitored at 500 nm (Fig. 4b, black line). The broad emission band was also observed for a chloroform solution at lower concentration (Fig. S1,†c = 1 × 10−6 M), suggesting that the long-wavelength tail is predominantly related to intramolecular excimers. The intramolecular exciton coupling and excimer formation significantly decreases the emission quantum yield Φ. Whereas the quantum yield of the linear reference compound 2 in chloroform solution is 0.90, it is reduced to 0.14 in the case of 1. Interestingly, Φ is still twice as large as that of cyclophane 3 (Φ = 0.07) under similar conditions.14
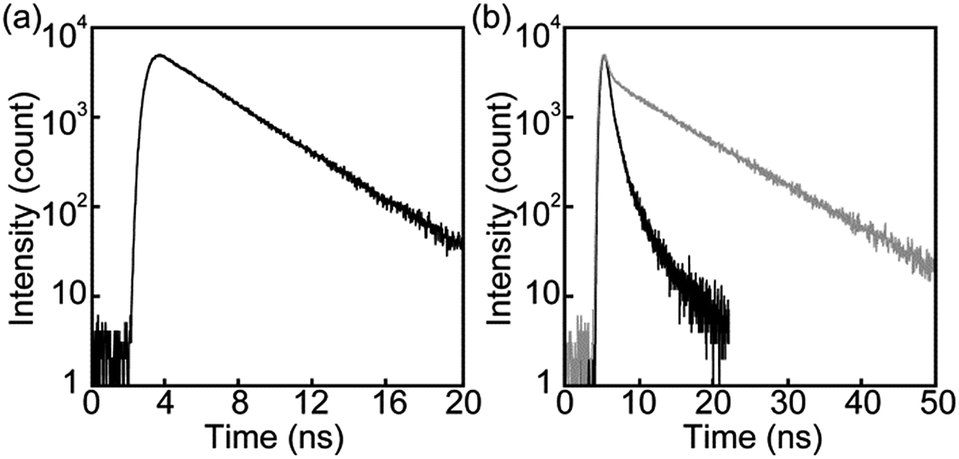
Steady-state photoluminescence spectra of 1 in the solid states were also measured at room temperature (Fig. 5). The emission band of the Y-form is structureless and displays only one maximum at 567 nm. Annealing the Y-form at 150 °C for 10 min results in a blue shift of the emission band (λem,max = 560 nm) and the band narrows a bit. The thermally induced change of the emission characteristics matches the photoluminescence color change from yellow to yellowish-green after the transition from the Y-form to the YG-form (Fig. 2). Emission decay profiles of the Y- and YG-form were measured to confirm that the emissive species contributing to each emission band are different from each other. Indeed, as shown in Fig. 6, the decay profiles are clearly different, but both could be fitted with tri-exponential decay functions. Whereas a long lifetime of 20 ns indicative of excimers was detected for the Y-form, no emissive species with such a long lifetime was observed for the YG-form (Table 1). When the YG-form was ground, the emission band was redshifted and broadened, which reflects the amorphous nature of the Am-form (vide infra). In the amorphous state, various arrangements of the emissive cores of 1 exist, and energy transfer can be expected to occur from higher energy sites to excimers. Annealing of the Am-form of 1 at 150 °C for 10 min affords an inhomogeneous solid that shows both green and yellow emission depending on the parts, which means cyclophane 1 forms several molecular arrangements. While compound 1 exhibits unambiguous emission color changes, the linear reference compound 2 never shows clear color changes in response to mechanical stimuli, though vibronic structure could be seen in the emission spectrum after grinding as shown in Fig. 7.
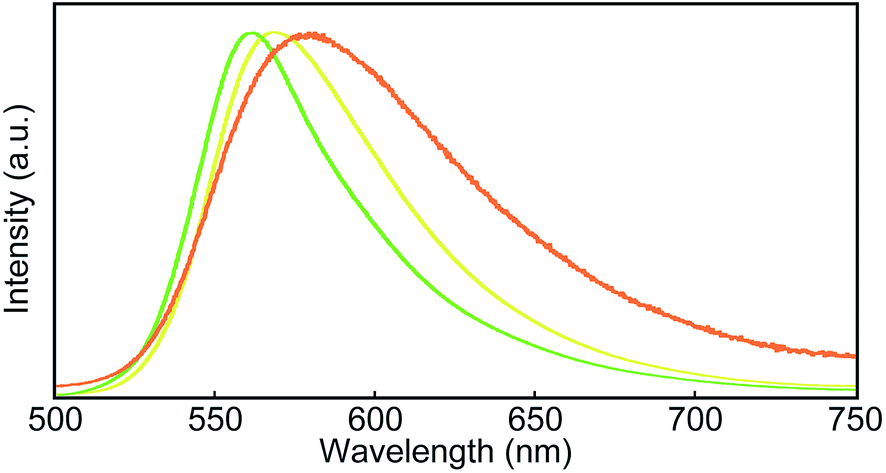 | ||
| Fig. 5 Photoluminescence spectra of the Y-form (yellow line), the YG-form (green line), and the ground YG-form (orange line) of cyclophane 1. All spectra were recorded at room temperature with λex = 400 nm. | ||
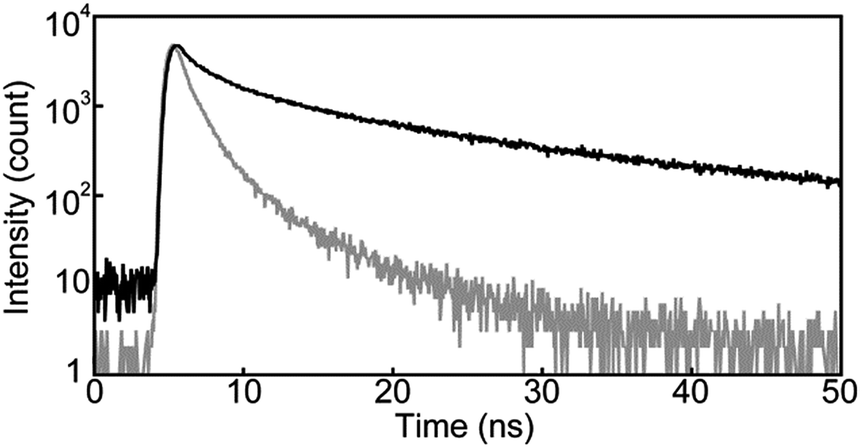 | ||
| Fig. 6 Emission decay profiles of the Y-form of cyclophane 1 collected at 600 nm (black line) and the YG-form of cyclophane 1 collected at 560 nm (grey line). All profiles were measured at room temperature with λex = 405 nm. | ||
| τ i (ns) | X 2 | Φ PL | |
|---|---|---|---|
| a All measurements were carried out at room temperature. b Monitored at 500 nm. c Monitored at 650 nm. d Monitored at 600 nm. e Monitored at 560 nm. | |||
| 1 in chloroformb | 0.2, 0.6, 2.2 | 1.09 | 0.14 |
| 1 in chloroformc | 0.5, 8.6 | 1.06 | |
| Y-Formd | 0.9, 4.8, 20 | 1.09 | 0.20 |
| YG-Forme | 0.4, 1.1, 3.9 | 1.01 | 0.14 |
| 2 in chloroformb | 3.1 | 1.03 | 0.90 |
 | ||
| Fig. 7 Photoluminescence spectra of the linear reference compound 2 in the solid state before (solid line) and after (dotted line) grinding. All spectra were recorded at room temperature with λex = 400 nm. | ||
To gain further insights into the color changing behavior, we investigated the phase behavior of the cyclophane 1 and the linear compound 2. Fig. 8 shows differential scanning calorimetry (DSC) curves (first heating) of the Y-form and the YG-form of 1 and the reference compound 2. Upon heating the Y-form, a weak exothermic peak appears at 116.3 °C, which is ascribed to a phase transition from the Y-form to the YG-form. The DSC curve also displays two endothermic peaks at 159.6 and 168.6 °C. The former peak is corresponding to a phase transition from the YG-form to another yellow emissive crystalline phase, whereas the latter peak is due to a phase transition to a LC phase (vide infra). Heating to above 250 °C leads to decomposition of 1 and no discrete clearing temperature could be discerned. Because the thermogravimetric analysis (TGA) trace of 1 (Fig. 9) shows no clear weight loss below 250 °C, the phase transition at 116.3 °C is not caused by the release of trapped solvent. The powder XRD patterns of cyclophane 1 confirm that the observed color change from yellow to yellowish-green (Y-form → YG-form) is ascribed to the change of the molecular assembled state. As shown in Fig. 10, the XRD patterns of both the Y- and YG-form show many peaks, indicative of their crystalline nature. However, the peak positions of the Y-form are clearly different from those of the YG-form, pointing to different crystal structures. As the peak observed in the DSC of the Y-form of 1 at 116.3 °C upon heating is exothermic, the transition from the Y-form to YG-form is a transition from thermodynamically metastable to stable state. Indeed, the YG-form shows no peaks in the DSC curve upon heating (Fig. 8, middle). In the previous study, cyclophane 3 having tetraethylene glycol as the bridge linkers gave one endothermic peak at 221 °C on heating until the compound shows decomposition, which coincides with the photoluminescence color change from yellow to reddish orange.14 Just elongation of linkers gives clear changes in the phase transition and color changing behavior.
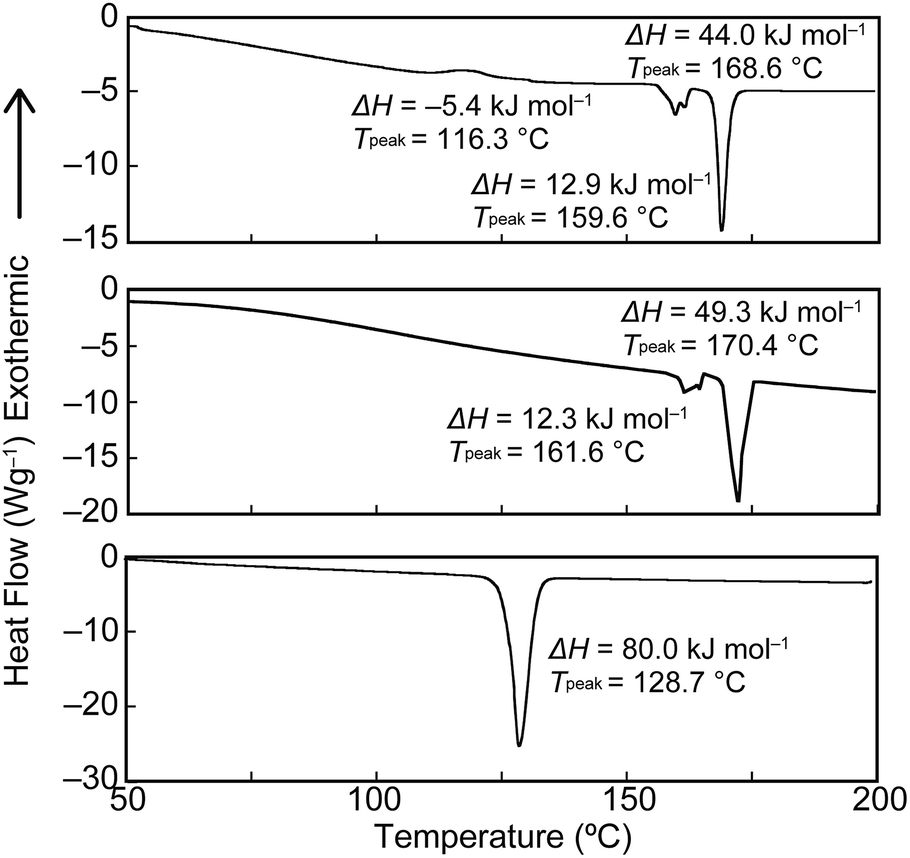 | ||
| Fig. 8 First heating DSC traces of the Y-form (top) and the YG-form (middle) of cyclophane 1, and the linear reference compound 2 (bottom). | ||
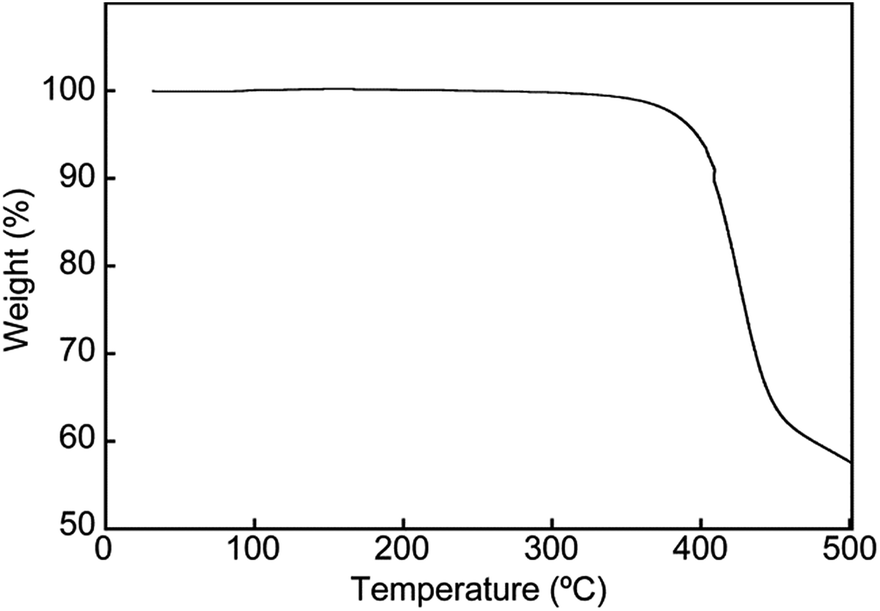 | ||
| Fig. 9 TGA curve acquired for the Y-form of cyclophane 1. The measurement was conducted under N2. | ||
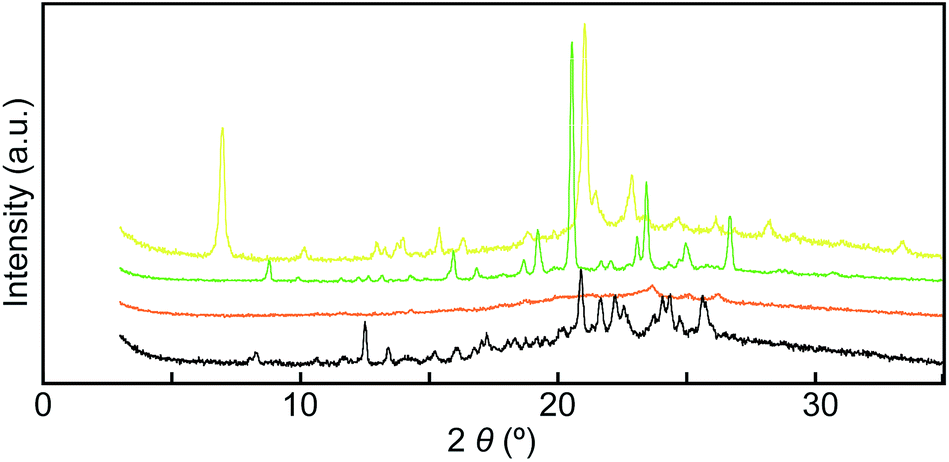 | ||
| Fig. 10 Powder X-ray diffraction patterns of cyclophane 1 in the Y-form (yellow line), the YG-form (green line), the Am-form (orange line), and the annealed Am-form (black line). All measurements were carried out at room temperature. | ||
Mechanical grinding significantly affects the XRD patterns of cyclophane 1. As shown in Fig. 10, all sharp peaks disappear in the XRD pattern for the Am-form that had been prepared by grinding the Y-form (Fig. 10, orange line), indicating that cyclophane 1 forms an amorphous solid after grinding. Subsequent annealing leads to the appearance of many peaks (Fig. 10, black line), but the diffraction pattern is different from those of the Y- and YG-forms. This result suggests that cyclophane 1 forms several other molecular assembled structures, because both yellow and green emissive parts were concomitantly obtained after thermal treatment (Fig. 2). Because one exothermic peak was observed on the DSC curve of the Am-form (Fig. S2†), the Am-form was thermodynamically metastable. In the case of cyclophane 3, annealing the ground sample recovers the original XRD pattern.14 Therefore, elongation of spacer chains allows compound 1 to display a more complicated phase transition behavior.
Moreover, compound 1 shows a LC phase above 168.6 °C, very much in contrast to the previously reported cyclophane 3 (ref. 14) and the linear reference compound 2. The XRD pattern measured at 190 °C (Fig. 11) shows no clear peaks, pointing to a nematic phase. This is supported by the very low viscosity and the schlieren texture observed in polarized optical microscopic images (Fig. 12). An LC texture was also reported for other cyclic compounds.25 It is noteworthy that the reference compound 2 displays no thermotropic behavior; indeed, the DSC trace shows only one endothermic peak corresponding to a transition from crystal to isotropic phase at 128.7 °C (Fig. 8, bottom). Thus, it appears that the cyclic structure of 1 restricts the stable and closed packing of molecules, resulting in the appearance of an LC phase.
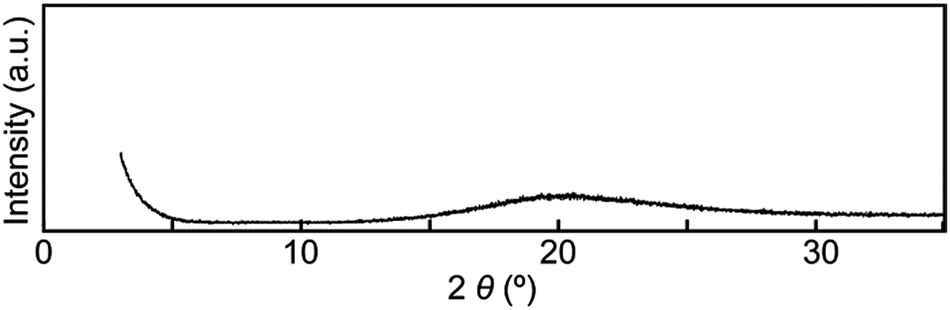 | ||
| Fig. 11 Powder X-ray diffraction pattern of cyclophane 1 at 190 °C. | ||
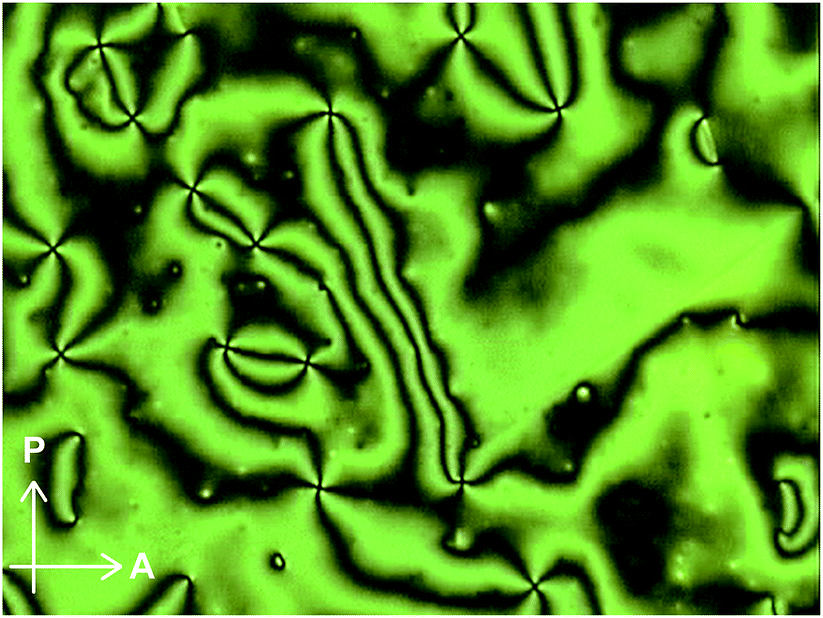 | ||
| Fig. 12 Polarized optical microscopic image of cyclophane 1 at 200 °C. | ||
Conclusions
In conclusion, our results suggest that the thermo- and mechanoresponsive luminescence behavior of cyclophanes is a general effect that should be broadly exploitable. Cyclophane 1 exhibits clear color changes upon mechanical grinding and exposure to heat, whereas the linear reference compound 2 shows no such changes. The stimuli-responsive behavior of the new compound is quite different than that of a previously reported cyclophane with similar structure but shorter bridges, which demonstrates that merely changing the ring size is allows one to change the stimuli-responsiveness and the phase behavior of luminescent cyclophanes in a significant manner. Our finding clearly confirms that the integration of luminescent motifs into cyclic structures is a promising approach to create luminescent compounds with stimuli-responsive properties in the solid states.Experimental section
All reagents and solvents were purchased from Aldrich and Tokyo Kasei. Unless otherwise noted, all reactions were carried out under nitrogen atmosphere. Silica gel column chromatography was carried out with silica gel from Kanto Chemicals (silica gel 60 N, spherical, 63–210 μm). 1H NMR spectra were recorded on a JEOL JNM-ECX 400 spectrometer and all chemical shifts are quoted on the δ-scale in ppm relative to the signal of tetramethylsilane (at 0.00) as an internal standard. Proton-decoupled 13C NMR spectra were recorded on a JEOL JNM-ECX 400 spectrometer and all chemical shifts (δ) are reported in ppm using residual solvent as the internal standard (CDCl3 at 77.16). Coupling constants (J) are reported in Hz and relative intensities are also shown. Elemental analysis was conducted with an Exeter Analytical CE440 Elemental Analyzer. Matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectra were measured on an AB SCIEX TOF/TOF 5800. The DSC measurements were carried out using a Rigaku Thermo plus EVO DSC8230 with heating/cooling rates of 10 °C min−1 under nitrogen atmosphere. The thermogravimetric analysis was conducted using a Rigaku Thermo plus TG8120 under nitrogen atmosphere. Powder X-ray diffraction experiments were conducted with a Rigaku SmartLab. An Anton Paar DHS1100 was attached to the Rigaku SmartLab for PXRD measurements at elevated temperature. UV-vis absorption spectra were recorded on a JASCO V-550. Steady-state fluorescence spectra were recorded on a JASCO FP-6500. The solid samples for steady-state fluorescence spectra were carefully sandwiched between quartz substrates and were set in the sample chamber of the spectrometer so that the detector gathers fluorescence from the surface of the sandwiched solid. Time-resolved fluorescence measurements were carried out with a Hamamatsu Photonics Quantaurus-Tau at room temperature. Quantum efficiencies were measured with a Hamamatsu Photonics Quantaurus-QY at room temperature. Optical microscopy was conducted with an Olympus BX-60 optical microscope equipped with a Sony DXC-950 3CCD camera and a Mettler Toledo FP 90 hot stage. Cyclophane 1 was simply placed on a glass substrate for this experiment, because the compound was observed to show a strong tendency for homeotropic alignment when sandwiched between glass substrates, leading to disappearance of the schlieren texture.Acknowledgements
We thank Prof. Y. Urano for emission lifetime measurements and quantum yield measurements and Y. Takahashi and Prof. S. Noro for DSC measurements and TGA. Y. S. acknowledges support from JSPS KAKENHI Grant Numbers JP16K17885 and JP16H00818. Y. S. also acknowledges support from the Asahi Glass Foundation. C. W. acknowledges support from the National Center of Competence in Research (NCCR) Bio-Inspired Materials, a research instrument of the Swiss National Science Foundation, the European Research Council (ERC-2011-AdG 291490-MERESPO), and the Adolphe Merkle Foundation.Notes and references
- (a) C. J. Brown and A. C. Farthing, Nature, 1949, 164, 915–916 CrossRef CAS; (b) H. Hopf, Isr. J. Chem., 2012, 52, 18–19 CrossRef CAS.
- Modern Cyclophane Chemistry, ed. R. Gleiter and H. Hopf, Wiley-VCH, Weinheim, 2004 Search PubMed.
- D. J. Cram and H. Steinberg, J. Am. Chem. Soc., 1951, 73, 5691–5704 CrossRef CAS.
- (a) D. J. Cram and J. M. Cram, Acc. Chem. Res., 1971, 4, 204–213 CrossRef CAS; (b) C. Seel and F. Vögtle, Angew. Chem., Int. Ed. Engl., 1992, 31, 528–549 CrossRef; (c) J. O. Jeppesen, M. B. Nielsen and J. Becher, Chem. Rev., 2004, 104, 5115–5131 CrossRef CAS PubMed; (d) P. G. Ghasemabadi, T. Yao and G. J. Bodwell, Chem. Soc. Rev., 2015, 44, 6494–6518 RSC.
- J.-M. Lehn, Science, 1993, 260, 1762–1763 CAS.
- (a) R. M. Izatt, K. Pawlak, J. S. Bradshaw and R. L. Bruening, Chem. Rev., 1995, 95, 2529–2586 CrossRef CAS; (b) F. P. Schmidtchen and M. Berger, Chem. Rev., 1997, 97, 1609–1646 CrossRef CAS PubMed; (c) A. Jasat and J. C. Sherman, Chem. Rev., 1999, 99, 931–967 CrossRef CAS PubMed; (d) E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed; (e) D. Ramaiah, P. P. Neelakandan, A. K. Nair and R. R. Avirah, Chem. Soc. Rev., 2010, 39, 4158–4168 RSC; (f) M. Xue, Y. Yang, X. Chi, Z. Zhang and F. Huang, Acc. Chem. Res., 2012, 45, 1294–1308 CrossRef CAS PubMed.
- (a) D. B. Amabilino and J. F. Stoddart, Chem. Rev., 1995, 95, 2725–2828 CrossRef CAS; (b) V. Balzani, M. Gomez-Lopez and J. F. Stoddart, Acc. Chem. Res., 1998, 31, 405–414 CrossRef CAS; (c) A. R. Pease, J. O. Jeppesen, J. F. Stoddart, Y. Luo, C. P. Collier and J. R. Heath, Acc. Chem. Res., 2001, 34, 433–444 CrossRef CAS PubMed; (d) A. H. Flood, Y. Liu and J. F. Stoddart, in Modern Cyclophane Chemistry, ed. R. Gleiter and H. Hopf, Wiley-VCH, Weinheim, 2005, pp. 485–518 Search PubMed; (e) I. Aprahamian, O. S. Miljanic, W. R. Dichtel, K. Isoda, T. Yasuda, T. Kato and J. F. Stoddart, Bull. Chem. Soc. Jpn., 2007, 80, 1856–1869 CrossRef CAS; (f) A. C. Fahrenbach, C. J. Bruns, D. Cao and J. F. Stoddart, Acc. Chem. Res., 2012, 45, 1581–1592 CrossRef CAS PubMed.
- (a) P. Čudić, M. Žinić, V. Tomišić, V. Simeon, J.-P. Vigneron and J.-M. Lehn, J. Chem. Soc., Chem. Commun., 1995, 1073–1075 RSC; (b) M.-P. Teulade-Fichou, J.-P. Vigneron and J.-M. Lehn, J. Chem. Soc., Perkin Trans. 2, 1996, 2, 2169–2175 RSC; (c) O. Baudoin, F. Gonnet, M.-P. Teulade-Fichou, J.-P. Vigneron, J.-C. Tabet and J.-M. Lehn, Chem.–Eur. J., 1999, 5, 2762–2771 CrossRef CAS.
- (a) P. P. Neelakandan and D. Ramaiah, Angew. Chem., Int. Ed., 2008, 47, 8407–8411 CrossRef CAS PubMed; (b) P. P. Neelakandan, K. S. Sanju and D. Ramaiah, Photochem. Photobiol., 2010, 86, 282–289 CrossRef CAS PubMed.
- (a) M. Inouye, K. Fujimoto, M. Furusyo and H. Nakazumi, J. Am. Chem. Soc., 1999, 121, 1452–1458 CrossRef CAS; (b) H. Abe, Y. Mawatari, H. Teraoka, K. Fujimoto and M. Inouye, J. Org. Chem., 2004, 69, 495–504 CrossRef CAS PubMed.
- L. Qiu, C. Zhu, H. Chen, M. Hu, W. He and Z. Guo, Chem. Commun., 2014, 50, 4631–4634 RSC.
- P. Spenst and F. Würthner, Angew. Chem., Int. Ed., 2015, 54, 10165–10168 CrossRef CAS PubMed.
- P. C. Nandajan, P. P. Neelakandan and D. Ramaiah, RSC Adv., 2013, 3, 5624–5630 RSC.
- Y. Sagara, Y. C. Simon, N. Tamaoki and C. Weder, Chem. Commun., 2016, 52, 5694–5697 RSC.
- (a) Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS PubMed; (b) K. Araki and T. Mutai, Photochemistry, 2016, 43, 191–225 CAS; (c) T. Seki and H. Ito, Chem.–Eur. J., 2016, 22, 4322–4329 CrossRef CAS PubMed; (d) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed; (e) A. P. Haehnel, Y. Sagara, Y. C. Simon and C. Weder, Top. Curr. Chem., 2015, 369, 345–375 CrossRef PubMed; (f) Z. Ma, Z. Wang, M. Teng, Z. Xu and X. Jia, ChemPhysChem, 2015, 16, 1811–1828 CrossRef CAS PubMed; (g) F. Ciardelli, G. Ruggeri and A. Pucci, Chem. Soc. Rev., 2013, 42, 857–870 RSC; (h) X. Zhang, Z. Chi, Y. Zhang, S. Liu and J. Xu, J. Mater. Chem. C, 2013, 1, 3376–3390 RSC; (i) Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC; (j) Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS PubMed.
- (a) A. Seeboth, D. Lötzsch, R. Ruhmann and O. Muehling, Chem. Rev., 2014, 114, 3037–3068 CrossRef CAS PubMed; (b) J. Lott, C. Ryan, B. Valle, J. R. Johnson, D. A. Schiraldi, J. Shan, K. D. Singer and C. Weder, Adv. Mater., 2011, 23, 2425–2429 CrossRef CAS PubMed; (c) S. Yamane, Y. Sagara and T. Kato, Chem. Commun., 2009, 3597–3599 RSC; (d) Y. Zhao, H. Gao, Y. Fan, T. Zhou, Z. Su, Y. Liu and Y. Wang, Adv. Mater., 2009, 21, 3165–3169 CrossRef CAS; (e) N. S. S. Kumar, S. Varghese, N. P. Rath and S. Das, J. Phys. Chem. C, 2008, 112, 8429–8437 CrossRef CAS; (f) T. Mutai, H. Tomoda, T. Ohkawa, Y. Yabe and K. Araki, Angew. Chem., Int. Ed., 2008, 47, 9522–9524 CrossRef CAS PubMed; (g) T. Mutai, H. Satou and K. Araki, Nat. Mater., 2005, 4, 685–687 CrossRef CAS PubMed; (h) A. Kishimura, T. Yamashita, K. Yamaguchi and T. Aida, Nat. Mater., 2005, 4, 546–549 CrossRef CAS PubMed; (i) R. Davis, N. P. Rath and S. Das, Chem. Commun., 2004, 74–75 RSC.
- (a) T. Seki, Y. Takamatsu and H. Ito, J. Am. Chem. Soc., 2016, 138, 6252–6260 CrossRef CAS PubMed; (b) Y. Sagara, A. Lavrenova, A. Crochet, Y. C. Simon, K. M. Fromm and C. Weder, Chem.–Eur. J., 2016, 22, 4374–4378 CrossRef CAS PubMed; (c) Z. Ma, Z. Wang, X. Meng, Z. Ma, Z. Xu, Y. Ma and X. Jia, Angew. Chem., Int. Ed., 2016, 55, 519–522 CrossRef CAS PubMed; (d) T. Seki, T. Ozaki, T. Okura, K. Asakura, A. Sakon, H. Uekusa and H. Ito, Chem. Sci., 2015, 6, 2187–2195 RSC; (e) H.-J. Kim, D. R. Whang, J. Gierschner, C. H. Lee and S. Y. Park, Angew. Chem., Int. Ed., 2015, 54, 4330–4333 CrossRef CAS PubMed; (f) S. Yagai, S. Okamura, Y. Nakano, M. Yamauchi, K. Kishikawa, T. Karatsu, A. Kitamura, A. Ueno, D. Kuzuhara, H. Yamada, T. Seki and H. Ito, Nat. Commun., 2014, 5, 4013 CAS; (g) R. Li, S. Xiao, Y. Li, Q. Lin, R. Zhang, J. Zhao, C. Yang, K. Zou, D. Li and T. Yi, Chem. Sci., 2014, 5, 3922–3928 RSC; (h) K. Nagura, S. Saito, H. Yusa, H. Yamawaki, H. Fujihisa, H. Sato, Y. Shimoikeda and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 10322–10325 CrossRef CAS PubMed; (i) T. Seki, K. Sakurada and H. Ito, Angew. Chem., Int. Ed., 2013, 52, 12828–12832 CrossRef CAS PubMed; (j) H. Ito, M. Muromoto, S. Kurenuma, S. Ishizaka, N. Kitamura, H. Sato and T. Seki, Nat. Commun., 2013, 4, 2009 Search PubMed; (k) W. Z. Yuan, Y. Q. Tan, Y. Y. Gong, P. Lu, J. W. Y. Lam, X. Y. Shen, C. F. Feng, H. H.-Y. Sung, Y. W. Lu, I. D. Williams, J. Z. Sun, Y. M. Zhang and B. Z. Tang, Adv. Mater., 2013, 25, 2837–2843 CrossRef CAS PubMed; (l) Y. Dong, B. Xu, J. Zhang, X. Tan, L. Wang, J. Chen, H. Lv, S. Wen, B. Li, L. Ye, B. Zou and W. Tian, Angew. Chem., Int. Ed., 2012, 51, 10782–10785 CrossRef CAS PubMed; (m) Y. Ren, W. H. Kan, V. Thangadurai and T. Baumgartner, Angew. Chem., Int. Ed., 2012, 51, 3964–3968 CrossRef CAS PubMed; (n) J. Wang, J. Mei, R. Hu, J. Z. Sun, A. Qin and B. Z. Tang, J. Am. Chem. Soc., 2012, 134, 9956–9966 CrossRef CAS PubMed; (o) M. Sase, S. Yamaguchi, Y. Sagara, I. Yoshikawa, T. Mutai and K. Araki, J. Mater. Chem., 2011, 21, 8347–8354 RSC; (p) G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160–2162 CrossRef CAS PubMed; (q) S.-J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M.-G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef CAS PubMed; (r) J. Kunzelman, M. Kinami, B. R. Crenshaw, J. D. Protasiewicz and C. Weder, Adv. Mater., 2008, 20, 119–122 CrossRef CAS; (s) Y. Sagara, T. Mutai, I. Yoshikawa and K. Araki, J. Am. Chem. Soc., 2007, 129, 1520–1521 CrossRef CAS PubMed.
- (a) J. Lott and C. Weder, Macromol. Chem. Phys., 2010, 211, 28–34 CrossRef CAS; (b) B. R. Crenshaw, M. Burnworth, D. Khariwala, A. Hiltner, P. T. Mather, R. Simha and C. Weder, Macromolecules, 2007, 40, 2400–2408 CrossRef CAS; (c) A. Pucci, F. Di Cuia, F. Signori and G. Ruggeri, J. Mater. Chem., 2007, 17, 783–790 RSC; (d) M. Kinami, B. R. Crenshaw and C. Weder, Chem. Mater., 2006, 18, 946–955 CrossRef CAS; (e) B. R. Crenshaw and C. Weder, Macromolecules, 2006, 39, 9581–9589 CrossRef CAS; (f) A. Pucci, M. Bertoldo and S. Bronco, Macromol. Rapid Commun., 2005, 26, 1043–1048 CrossRef CAS; (g) B. R. Crenshaw and C. Weder, Chem. Mater., 2003, 15, 4717–4724 CrossRef CAS; (h) C. Löwe and C. Weder, Adv. Mater., 2002, 14, 1625–1629 CrossRef.
- (a) M. Mitani, S. Ogata, S. Yamane, M. Yoshio, M. Hasegawa and T. Kato, J. Mater. Chem. C, 2016, 4, 2752–2760 RSC; (b) M. Mitani, S. Yamane, M. Yoshio, M. Funahashi and T. Kato, Mol. Cryst. Liq. Cryst., 2014, 594, 112–121 CrossRef CAS; (c) S. Yamane, Y. Sagara, T. Mutai, K. Araki and T. Kato, J. Mater. Chem. C, 2013, 1, 2648–2656 RSC; (d) Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2011, 50, 9128–9132 CrossRef CAS PubMed; (e) Y. Sagara, S. Yamane, T. Mutai, K. Araki and T. Kato, Adv. Funct. Mater., 2009, 19, 1869–1875 CrossRef CAS; (f) V. N. Kozhevnikov, B. Donnio and D. W. Bruce, Angew. Chem., Int. Ed., 2008, 47, 6286–6289 CrossRef CAS PubMed; (g) Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2008, 47, 5175–5178 CrossRef CAS PubMed.
- (a) Y. Sagara, T. Komatsu, T. Ueno, K. Hanaoka, T. Kato and T. Nagano, J. Am. Chem. Soc., 2014, 136, 4273–4280 CrossRef CAS PubMed; (b) Y. Sagara, T. Komatsu, T. Terai, T. Ueno, K. Hanaoka, T. Kato and T. Nagano, Chem.–Eur. J., 2014, 20, 10397–10403 CrossRef CAS PubMed; (c) Y. Sagara, T. Komatsu, T. Ueno, K. Hanaoka, T. Kato and T. Nagano, Adv. Funct. Mater., 2013, 23, 5277–5284 CrossRef CAS.
- T. Hirose and K. Matsuda, Chem. Commun., 2009, 5832–5834 RSC.
- (a) W. Wang, L. Wang, B. J. Palmer, G. J. Exarhos and A. D. Q. Li, J. Am. Chem. Soc., 2006, 128, 11150–11159 CrossRef CAS PubMed; (b) J. Feng, Y. Zhang, C. Zhao, R. Li, W. Xu, X. Li and J. Jiang, Chem.–Eur. J., 2008, 14, 7000–7010 CrossRef CAS PubMed; (c) F. Schlosser, M. Moos, C. Lambert and F. Würthner, Adv. Mater., 2013, 25, 410–414 CrossRef CAS PubMed; (d) K. E. Brown, W. A. Salamant, L. E. Shoer, R. M. Young and M. R. Wasielewski, J. Phys. Chem. Lett., 2014, 5, 2588–2593 CrossRef CAS PubMed.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CrossRef CAS.
- J. B. Birks, Rep. Prog. Phys., 1975, 38, 903–974 CrossRef CAS.
- (a) P. R. Ashton, D. Joachimi, N. Spencer, J. F. Stoddart, C. Tschierske, A. J. P. White, D. J. Williams and K. Zab, Angew. Chem., Int. Ed. Engl., 1994, 33, 1503–1506 CrossRef; (b) B. Neumann, D. Joachimi and C. Tschierske, Adv. Mater., 1997, 9, 241–244 CrossRef CAS; (c) B. Neumann, T. Hegmann, C. Tschierske, B. Neumann and R. Wolf, Chem. Commun., 1998, 105–106 RSC; (d) B. Neumann, T. Hegmann, C. Wagner, P. R. Ashton, R. Wolf and C. Tschierske, J. Mater. Chem., 2003, 13, 778–784 RSC; (e) T. Hegmann, B. Neumann, R. Wolf and C. Tschierske, J. Mater. Chem., 2005, 15, 1025–1034 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic schemes and additional absorption and fluorescence spectra. See DOI: 10.1039/c6ra18348d |
| This journal is © The Royal Society of Chemistry 2016 |