
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, crystal structure and hydrolysis of novel isomeric cage (P–C/P–O)-phosphoranes on the basis of 4,4,5,5-tetramethyl-2-(2-oxo-1,2-diphenylethoxy)-1,3,2-dioxaphospholane and hexafluoroacetone†
Nadezhda R.
Khasiyatullina
a,
Vladimir F.
Mironov
*ab,
Dmitry B.
Krivolapov
a,
Ekaterina V.
Mironova
ab and
Oleg I.
Gnezdilov
c
aA. E. Arbuzov Institute of Organic and Physical Chemistry, Russian Academy of Sciences, Arbuzov Str. 8, 420088 Kazan, Russia. E-mail: mironov@iopc.ru
bKazan (Volga Region) Federal University, Kremlyovskaya Str. 18, Kazan, 420008, Russia
cE. K. Zavoisky Kazan Physical-Technical Institute, Russian Academy of Sciences, Sibirskiy Trakt 10/7, Kazan, 420029, Russia
First published on 25th August 2016
Abstract
The reaction of 4,4,5,5-tetramethyl-2-(2-oxo-1,2-diphenylethoxy)-1,3,2-dioxaphospholane with hexafluoroacetone leads to the simultaneous formation of regioisomeric cage (P–C/P–O)-phosphoranes, the structures of which are unequivocally confirmed by XRD. The rearrangement of the P–C-isomer to P–O-isomer with high stereoselectivity (>96%) takes place in methylene chloride solution with the retention of the phosphorus coordination. It was found that the stepwise hydrolysis of the P–O-isomer initially gives 2-(2,3-dihydroxy-1,2-diphenyl-3-trifluoromethyl-4,4,4-trifluorobutyloxy)-4,4,5,5-tetramethyl-2-oxo-1,3,2-dioxaphospholane as the only stereoisomer whose structure is also confirmed by XRD. Further hydrolysis of this compound leads to the formation of 2,3-dihydroxy-3-trifluoromethyl-4,4,4-trifluoro-1,2-diphenylbutylphosphate and pinacol, which forms the solvate in the crystal. Hydrolysis of the P–C-isomer yields 2-hydroxy-4,4,5,5-tetramethyl-2-oxo-1,3,2-dioxaphospholane, benzoin and hexafluoroisopropanol.
Introduction
Pentacoordinated phosphorus compounds are key intermediates in phosphoryl group transfer reactions, which are important in the processes of cell viability1–7 and in the origin and development of life.8 Such phosphorus derivatives are intermediates in the nucleophilic substitution reactions at the tetrahedral phosphorus atom,9–13 among which the most important for organic synthesis are the Wittig,14 Appel,15,16 and Mitsunobu17–19 reactions, which are well described. Therefore, the synthesis, structure and chemical transformations of phosphoranes have attracted considerable attention.20–27 Among the diverse synthetic methods for the preparation of phosphoranes, several general approaches based on the addition reactions of P(III)-derivatives to unsaturated systems, various reactions of tetracoordinated phosphorus and substitution reactions at P(V)28,29 should be noted.Recently, we developed a new approach for the preparation of phosphoranes based on the cascade reactions of P(III)-derivatives, containing an unsaturated moiety with carbonyl compounds, which leads to P(V)–C cage heterocycles.30–34Scheme 1, which shows the synthetic possibilities of this approach, is an example of the reactions of benzodioxaphosphole derivatives 1 with hexafluoroacetone. It is assumed that the reactions proceed through intermediate P+–C–O− bipolar ions followed by the transfer of the reactive center on the exocyclic unsaturated substituent, which lead to the formation of the corresponding cage phosphoranes 2–4 bearing the P–C-bond.30–32
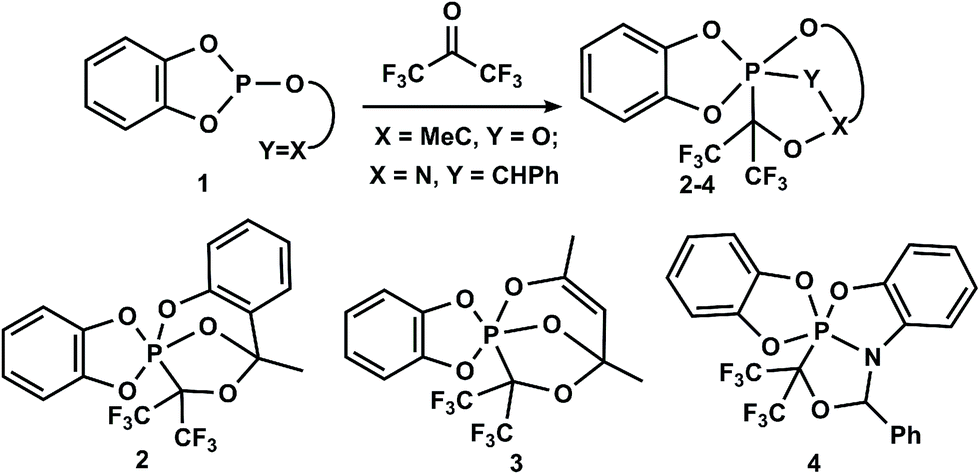 | ||
Scheme 1 Use of P(III)-derivatives bearing an exocyclic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or CN bond in the synthesis of cage phosphoranes. O or CN bond in the synthesis of cage phosphoranes. | ||
Scheme 2 demonstrates the synthetic potential of the intramolecular cascade cyclization of P(III)-derivatives 1 under the action of prochiral trifluoropyruvic acid ethyl ester and chloral, which allows the P–C-cage phosphoranes 5–8 to be obtained with high stereoselectivity.33,34
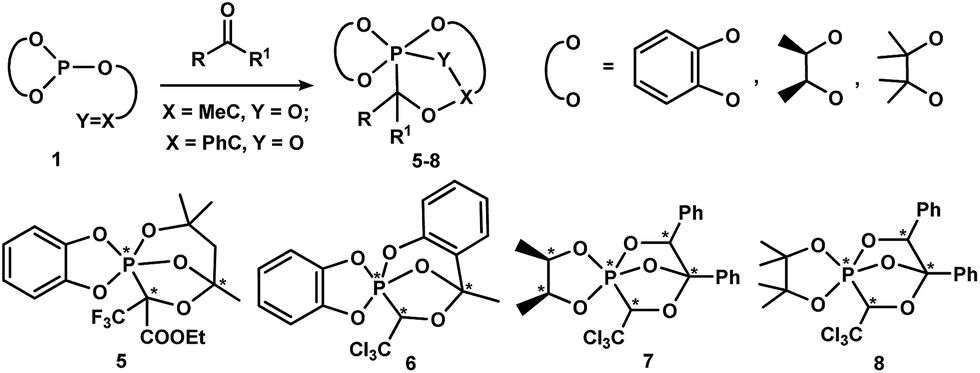 | ||
| Scheme 2 Reaction of P(III)-derivatives 1 bearing an exocyclic CO bond with trifluoropyruvic acid ethyl ester and chloral. | ||
None of these reactions afford pentaalkoxyphosphoranes, the products of intramolecular PCO/POC-rearrangement, which is characteristic to the reaction of ordinary trialkylphosphites with the carbonyl compounds mentioned above.35,36
Recently, we have shown37 that the inclusion of a phosphorus(III) atom in the dioxaphospholane cycle results in the simultaneous formation of PCO- and POC-isomers (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) of cage phosphoranes10,11 in the reaction of 4,5-dimethyl-2-(2-oxo-1,2-diphenylethoxy)-1,3,2-dioxaphospholane9 with hexafluoroacetone (Scheme 3). PCO-phosphorane 10 is subjected to intramolecular PCO/POC-rearrangement during storage (CH2Cl2, 20 °C, 30 days) and yields the POC-species 11.
:1) of cage phosphoranes10,11 in the reaction of 4,5-dimethyl-2-(2-oxo-1,2-diphenylethoxy)-1,3,2-dioxaphospholane9 with hexafluoroacetone (Scheme 3). PCO-phosphorane 10 is subjected to intramolecular PCO/POC-rearrangement during storage (CH2Cl2, 20 °C, 30 days) and yields the POC-species 11.
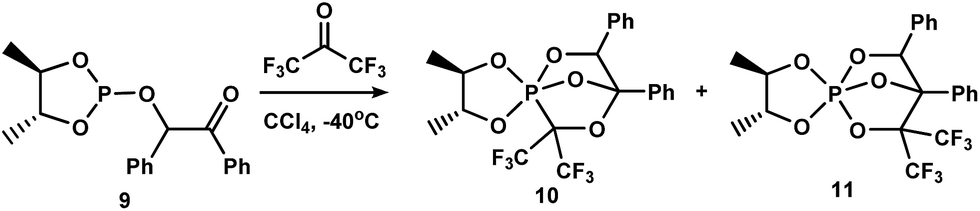 | ||
| Scheme 3 Simultaneous formation of PCO- and POC-isomers 10 and 11 in the reaction of phospholane 1 with hexafluoroacetone. | ||
Results and discussion
Considering that the related PCO/POC-rearrangement in a series of 1-hydroxyalkylphosphonates (–phosphinates) is facilitated not only by electron-withdrawing substituents at the carbon atom bonded with OH-group, but also electron-donor substituents at the phosphorus atom,38 we introduced 4,4,5,5-tetramethyl-2-(2-oxo-1,2-diphenylethoxy)-1,3,2-dioxaphospholane 12 in the reaction with hexafluoroacetone. Tetramethyl-substituted dioxaphospholane 12 has essentially more electron donor cyclic moieties as compared with the dimethyl-substituted phospholane 9, which, however, also effectively stabilizes the phosphorus pentacoordinated state. Compound 12 was obtained by the phosphorylation of benzoin with 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane in the presence of triethylamine according to the data.39The reaction of phosphite 12 with hexafluoroacetone proceeds in mild conditions (CCl4, −40 °C) with the simultaneous formation of two isomeric pentacoordinated phosphorus species 13 and 14 (Scheme 4), which have two high-field signals at δP −24.5 and δP −26.5 ppm in a ratio of 10:1 in their 31P–{1H} NMR spectra (a day after the reaction). The molecular ion peaks for compounds 13 and 14 are identical (ESI, m/z 523.9) and correspond to the reaction products with a composition of 1:1, and their fragmentation is not significantly different. After the removal of the solvent, the oily residue crystallizes with the formation of compound 13 during storage. It manifests itself as a doublet in the high-field region (CCl4, δP −25.1 ppm, 3JPCCH = 15.6 Hz) of the 31P–{1H} NMR spectrum. Taking into account the spectral data (see ESI†), we determined the structure of compound 13 to be 1-(2,3-butylenedioxy)-6,6-bis(trifluoromethyl)-3,4-diphenyl-1,2,5,7-phosphatrioxabicyclo[2.2.11,4]heptane. The 19F NMR spectrum contains two signals at δF −68.48 (qd, 4JFCCCF = 10.3 Hz, 3JPCCF = 4.6 Hz) and δF −68.94 ppm (qd, 4JFCCCF = 10.3 Hz, 3JPCCF 2.3 Hz) with an equal integral intensity, which correspond to the non-equivalent fluorine atoms of CF3-groups. In contrast to the abovementioned phosphite 9, which is a hexafluoroacetone system, the formation of PC-phosphorane 13 is a kinetically preferred process.
 | ||
| Scheme 4 The reaction of phospholane 12 with hexafluoroacetone. The same numbering of the atoms in structures 13–17 is given for convenience. | ||
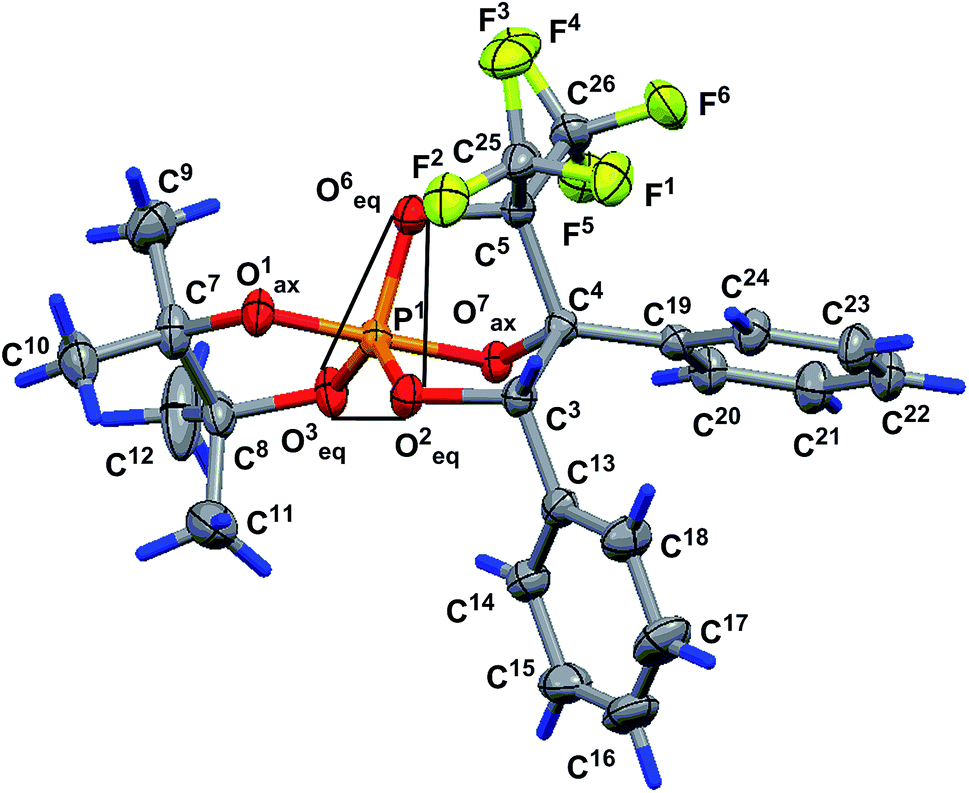
The structure of phosphorane 13 was confirmed by single crystal XRD. The geometry of molecule 13 in the crystal (conglomerate) is shown in Fig. 1, and the main geometrical parameters (bond lengths and bond and torsion angles) are listed in the figure caption. The configuration of the chiral atoms is PR1CR3CS4. A small deviation in the phosphorus atom from the O3O2O6 plane [by −0.114(2) Å] allows us to conclude that the phosphorus atom has a slightly distorted trigonal bipyramidal configuration with a planar 0.085(2) Å base, formed by the P1, O2, O3 and C6 atoms. The O1 and O7 atoms located in the apical positions deviate from this plane by −1.696 (5) and 1.619(5) Å, respectively [bond angle of Î1–P1–O7 is equal to 172.4 (3°)]. One of the two trifluoromethyl groups (C25F3) is almost in this plane [C25 atom deviation from it is 0.21(1) Å]. The O1–P1–O3, O1–P1–O2, O1–P1–C6, O2–P1–O7, O7–P1–C6 and O3–P1–O7 bond angles vary within the limits of 83.3(3)–96.9(3)°. The O2–P1–C6, O2–P1–O3 and O3–P1–C6 bond angle sum in the pyramid base is equal to 358.5(2)° and points to a trigonal bipyramidal phosphorus configuration close to an almost regular configuration. The P1–O3 equatorial bond length is slightly smaller [1.583(5) Å] than the P1–O1 and P1–O7 axial bond lengths of [1.613(3) and 1.711(5) Å].
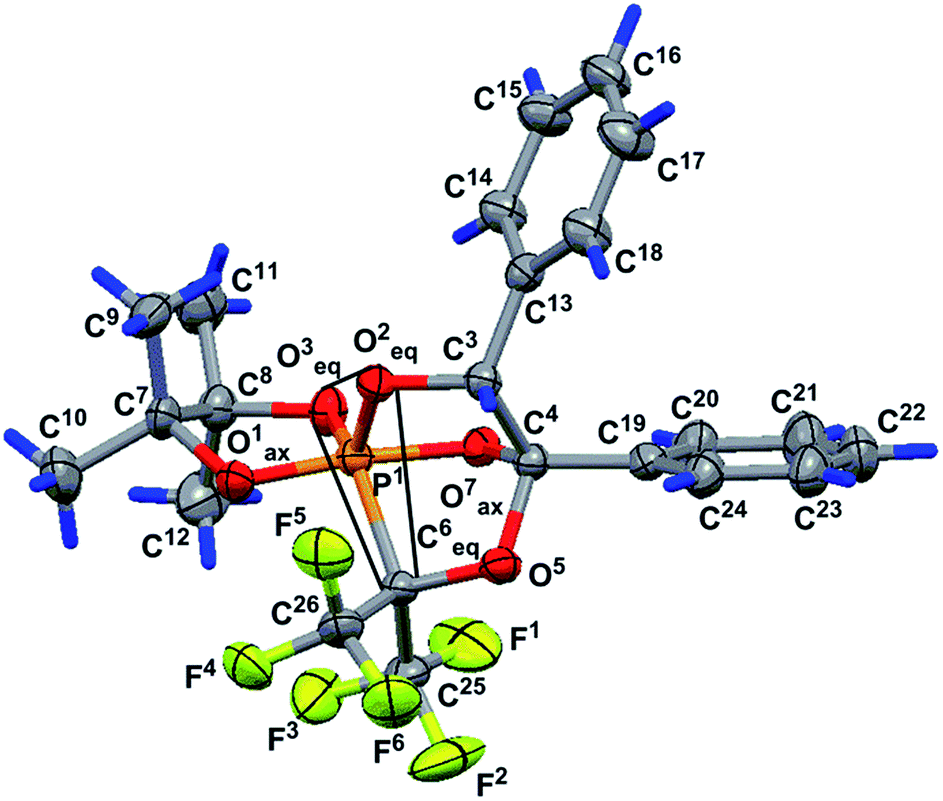 | ||
| Fig. 1 Geometry of molecule 13 in a crystal (conglomerate). Selected bond lengths (Å), and bond and torsion angles (°): P1–O1 1.613(5), P1–O2 1.619(5), P1–O3 1.583(5), P1–O7 1.711(5), P1–C6 1.930(7), O1–P1–O2 96.3(3), O1–P1–O3 92.7(3), O1–P1–O7 172.4(3), O1–P1–C6 92.8(3), O2–P1–O3 120.9(3), O2–P1–O7 90.3(2), O2–P1–C6 100.7(3), O3–P1–O7 85.7(2), O3–P1–C6 136.9(3), O7–P1–C6 83.3(3), C9–C7–C8–C12 −161.4(8), P1–O7–C4–C19 177.5(5), C13–C3–C4–C19 46.2(8) and O7–C4–C19–C20 1(1). Hereinafter, non-hydrogen atoms are shown in view of thermal ellipsoids with a probability of 30%, and the trigonal pyramid base is outlined with thin lines. | ||
The concurrence of the P1–O1 and P1–O7 axial bond length sum [3.324(5) Å] and O1⋯O7 distance [3.317(7) Å] can be considered as evidence of the regular trigonal bipyramidal phosphorus configuration. The P1–C6 bond length is equal to 1.930(7) Å. A five-membered dioxaphospholane cycle occupies the axial-equatorial position in the trigonal bipyramid (O1 is axial and O3 is equatorial), its conformation is envelope, where four atoms, O1, P1, O3 and C8, lie in one plane [planar within 0.004(5) Å], and the C7 atom deviates from this plane by 0.518(9) Å. The C9 and C12 atoms deviate from this plane by 2.05(1) and −1.43(1) Å, respectively, and they occupy axial positions in the cycle. The C10 and C11 atoms also deviate from the O1P1O3C8 plane by −0.05(1) and 0.91(1) Å, respectively, and are located in equatorial positions. The O2 and C6 atoms deviate in opposite sides from the O1P1O3C7 plane [their deviations are 1.377(5) and −1.297(8) Å, respectively] and occupy equatorial and axial positions in the five-membered cycle. The C10 and O7 deviations are minimal [they deviate by −0.05(1) and −0.213(5) Å, respectively] and we can assume that they lie in the O1P1O3C8 plane.
The O1P1O3C8 fragment can also be considered as a part of the most elongated seven-membered approximately planar C8O3P1O1O7C4C19 system [planar within 0.135(5) Å]. The plane of the phenyl substituent in the fourth position of the bicycloheptane 13 is slightly turned in the dihedral angle of 6.2(5)° relative to the seven-membered fragment. The conformation of the five-membered P1O2C3C4O7 cycle of molecule 13 is envelope (Fig. 1). It includes a four-membered plane P1O2C3C4 fragment [planar within 0.025(5) Å], and the O7 atom deviates from this plane by 0.760(5) Å. The O1, O3, O5, C6, C13 and C19 atoms deviate from the abovementioned fragment by the values of −0.742(5), 1.302(5), −1.391(5), −1.574(7), 1.257(7) and 0.355(3) Å, respectively. This is due to fact that the C13 and C19 atoms deviate to one side point out of the cis-orientation of the phenyl substituents in the five-membered cycle discussed above. The conformation of the other five-membered P1C6O5C4O7 ring of the rigid bicycloheptane scaffold is a slightly distorted envelope. The four-membered P1C6O5C4 fragment is planar within 0.066(5) Å, and the O7 atom deviates from this plane by −0.853(5) Å. The O1, O2, O3, C3, C19, C25 and C26 atoms deviate from the abovementioned fragment by the values of −0.699(5), 1.392(5), −0.949(5), 1.337(7), −0.579(7), −1.40(1) and 1.097(9) Å, respectively.
During storage in very polar dichloromethane at 20 °C for about five days, compound 13 underwent a gradual conversion to compound 14 (after four days, the ratio of compounds 13/14 was 1 to 20). Fig. 2 shows the spectral image of this process according to 31P–{1H} NMR. The figure shows that phosphorane 13 (CH2Cl2, δP −25.0 ppm) was completely converted to pentaoxyphosphorane 14 (CH2Cl2, δP −26.7 ppm), which is a minor compound in the reaction in tetrachloromethane. This compound was isolated in the individual state by crystallization under a layer of pentane and characterized by spectral methods, which allowed the assignment of the structure of 1-(2,3-butylenedioxy)-5,5-bis(trifluoromethyl)-3,4-diphenyl-1,2,6,7-phosphatrioxabicyclo[2.2.11,4]heptane 14, the product of PCO/POC-rearrangement. In the 13C–{1H} NMR spectrum of compound 14, the carbon atom bonded to CF3-groups resonates at 83.41 ppm as a septet with coupling through two bonds from fluorine (2JCCF = 29.3 Hz), unlike the spectrum of compound 13 in which the oxygenated carbon in the OC(CF3)2 fragment manifests itself as a doublet of septets at 77.78 ppm with direct spin–spin coupling from phosphorus (1JPC = 154.8 Hz, 2JFCC = 30.7 Hz). The signals of the CF3-groups in the 19F NMR spectrum of compound 14 appear as two quartets at δF −68.22 ppm and −72.35 ppm (4JFCCCF = 9.5 Hz).
 | ||
| Fig. 2 Conversion of P–C-phosphorane 13 to P–O-species 14 according to 31P–{1H} NMR spectra (CH2Cl2, in one day, and after two, three and five days). | ||
Its structure was also confirmed by single crystal XRD. Fig. 3 presents the geometry of the molecule in a crystal (the main geometrical parameters, including bond length, valence and torsion angles of the molecule above, are presented in the ESI file†). The configuration of the chiral atoms is PR1CR3CS4/PS1CS3CR4 (racemate). The meaningless deviation of the phosphorus atom from the O3O2C6 plane by −0.0580(7) Å allows to conclude that the geometry of phosphorus in molecule 14 is a slightly distorted trigonal bipyramid with a planar base [within 0.043(2) Å], where the P1, O2, O3, C6 atoms are located. The O1 and O7 atoms located in apical positions deviate from the above plane by the values of −1.625(2) and 1.700(2) Å [the valence angle of O1–P1–O7 is equal to 177.5(1)°]. The values of the valence angles O1–P1–O3, O1–P1–O2, O1–P1–O6, O2–P1–O7, O6–P1–O7 and O3–P1–O7 vary within the limits of 86.1(1)–92.4(1)°, and the sum of the valence angles of O2–P1–O6, O2–P1–O3 and O3–P1–O6 at the pyramid base is 359.6(2)°, which also indicate the trigonal-bipyramidal configuration of the phosphorus atom close to the regular configuration. The equatorial P1–O2, P1–O3 and P1–O6 bonds [1.606(2), 1.582(2), and 1.647(2) Å, respectively] are slightly shorter than the axial P1–O1 and P1–O7 bonds [1.625(2) and 1.700(2) Å, respectively]. The fact of practical coincidence of the P1–O1 and P1–O7 axial bond values sum [3.325(5) Å] and O1⋯O7 distance [3.323(7) Å] is in favor of the regular trigonal bipyramid configuration. The five-membered tetramethyldioxaphospholane cycle occupies the axial-equatorial position in the trigonal bipyramid (O1 atom is axial and O3 atom is equatorial). Its conformation is a distorted envelope, where four O1, P1, O3 and C7 atoms lie in one plane [planar within 0.0482(7) Å], and the C8 atom deviates from the above plane by 0.422(4) Å. The C9 and C12 atoms deviate from the plane to the opposite sides by the rather significant distances of −1.525(5) and 1.963(6) Å, respectively (they occupy axial positions in the cycle), and the C10 and C11 atoms also deviate by the different values of 0.758(5) and −0.178(6) Å (they occupy equatorial positions in the cycle). The O2 and O6 atoms deviate to opposite sides from the O1P1O3C7 plane and occupy equatorial and axial positions in the five-membered cycle [their deviations are 1.182(2) and −1.406(2) Å, respectively]. The O7 atom deviation is minimal [−0.213(5) Å]; thus, it can be concluded that this atom lies in the O1P1O3C7 plane. Moreover, it should be noticed that the O1P1O3C7 fragment is turned to be a part of the more extended planar moiety of C7O3P1O1O3O7C4C19 in the molecule [planar within 0.054(4) Å]. The plane of the phenyl substituent position 4 (C19–24) is slightly turned to this seven-membered fragment [the dihedral angle between the corresponding planes is larger than the angle in molecule 13 and is equal to 9.2(2)°]. The conformation of the five-membered P1O2C3C4O7 cycle of the rigid bicycloheptane system of molecule 14 is an envelope [it contains a planar four-membered P1O2C3C4 fragment within 0.033(2) Å, and the O7 atom deviates from it by 0.806(2) Å]. The Î1, O3, C5, O6, C13 and C19 atoms deviate from the abovementioned planar fragment by values of −0.726(2), 1.224(2), −1.428(3), −1.341(2), 1.276(3) and 0.485(3) Å, respectively. The fact that the C13 and C19 atoms deviate to one side suggests the cis-orientation of the phenyl substituents in the abovementioned five-membered cycle. The conformation of the other five-membered P1O6C5C4O7 cycle of the rigid bicycloheptane system of molecule 14 is a slightly distorted envelope [four-membered P1O6C5C4 fragment is planar within 0.044(3) Å, and the O7 atom deviates from the above fragment by −0.851(2) Å]. The O1, O2, O3, C3, C19, C25 and C26 atoms deviate from the abovementioned planar fragment by the values of 0.804(2), 1.364(2), −1.078(2), 1.322(3), −0.576(3), 1.366(4) and −1.151(4) Å.
 | ||
| Fig. 3 Geometry of molecule 14 in a crystal (racemate, PS1CS3CR4-enantiomer is shown). The base of the trigonal bipyramid is shown by thin lines. Selected bond lengths (Å) and bond and torsion angles (°): P1–O1 1.625(2), P1–O2 1.606(2), P1–O3 1.582(2), P1–O6 1.647(2), P1–O7 1.700(2), O1–P1–O2 92.1(1), O1–P1–O3 92.4(1), O1–P1–O7 177.5(1), O1–P1–O6 91.6(1), O2–P1–O3 126.4(1), O2–P1–O7 90.4(1), O2–P1–O6 105.8(1), O3–P1–O7 86.1(1), O3–P1–O6 127.4(1), O6–P1–O7 87.8(1), C9–C7–C8–C11 −164.4(4), P1–O7–C4–C19 178.8(2), C13–C3–C4–C19 −40.9(3) and O7–C4–C19–C20 10.0(4). | ||
Considering that compounds 13 and 14 are formed simultaneously under very mild conditions (−40 °C) and the rearrangement of P–C-isomer 13 into P–O-isomer 14 is slow, it can be assumed that they are formed from the common unstable phosphorane intermediate A, which contains an oxaphosphirane cycle (Scheme 4). This intermediate is a product of the symmetry allowed [1 + 2]-cycloaddition reaction of phosphorus to the double bond of hexafluoroacetone. At low temperature, the cleavage of the three-membered ring occurs readily in two directions, I and II, which finally results in the formation of compounds 13 and 14. The direction I, which includes P–O bond cleavage and the formation of intermediate bipolar ions B and C, is a kinetically controlled and reversible process. Direction II, which includes P–C bond cleavage and the formation of intermediate bipolar ions D and E, irreversibly results in a thermodynamically controlled reaction product, the P–O-isomer 14. Thus, the driving force of the PCO/POC-rearrangements seems to be the greater thermodynamic stability of the resulting pentaalkoxyphosphorane 14 in comparison with its P–C-analogue 13. Furthermore, oxygen is more electronegative than carbon, and it is important for the stabilization of the phosphorus trigonal bipyramid, which increases its stability when acceptors are introduced to the phosphorus atom.
Due to the fact that racemic benzoin was used in the synthesis of phospholane 12 and in the reaction with hexafluoroacetone another chiral carbon (C4) formation occurred, two P–C diastereoisomers of isomer 13 should be formed. The formation of only one diastereoisomer indicates the high stereoselectivity of the second chiral center (C4) formed, which is probably due to the rigid spatial requirements for the attack of the alkoxide-anion to the carbonyl group of the bipolar ion B. The very important fact that the relative configuration of the chiral C3 and C4 atoms in the P–C- and P–O-isomers 13 and 14, according to XRD data, is the same indicates the highly stereoselective nature of the intramolecular PCO/POC-rearrangement.
Hydrolysis of P–C-isomer 13 leads to the formation of dioxaphospholane 15, benzoin, and hexafluoroisopropanol. Benzoin and compound 15 were isolated by crystallization of the reaction mixture from diethyl ester. The structures of benzoin and hexafluoroisopropanol were proven by comparison of their spectral characteristics (1H, 13C and 19F NMR) with the literature.40–43 The structure of dioxaphospholane 15 was established based on the comparison of its spectral parameters with that described in the literature44,45 and XRD.
The geometry of the molecule in a crystal (solvate with one water molecule) is presented in Fig. 4. The five-membered cycle of molecule 15 has an envelope conformation, accordingly with a planar O1P1O3C8 fragment within 0.115(4) Å, and the C7 atom deviates from the abovementioned plane by −0.492(8) Å. The O2, C9 and C12 atoms are located in axial positions (they deviate from the O1P1O3C8 plane by 1.574(5), −1.990(8) and 1.552(8) Å, respectively). The O4, C10 and C11 atoms are located in equatorial positions and deviate from the O1P1O3C8 plane by −0.851(4), 0.20(1) and −0.713(9) Å, respectively. Due to the presence of solvent water molecules in the crystal of 15, a classical hydrogen bond system is realized with the participation of the phosphoryl O4 oxygen and water molecule protons [O1S–H1S⋯O4-interaction, the parameters are O1S–H1S 0.72(9) Å, H1S⋯O4 2.0(1) Å, O1S⋯O4 2.75(1) Å, ∠O1S–H1S⋯O4 178(8)°, and symmetry operation −1/2 + x, 1/2 − y, 1 − z] and also between hydrogen at the O2 atom and water oxygen O1S [O2–H2⋯O1S-interaction, the parameters are O2–H2 0.86(7) Å, H2⋯O1S 1.61(7) Å, O2⋯O1S 2.46(1) Å, ∠O2–H2⋯O1S 168(9)°, and symmetry operation 1 + x, y, z]. Using the classical hydrogen interactions, the molecules in the crystal 15 form ribbons along the 0a crystallographic axis (see Fig. 5).
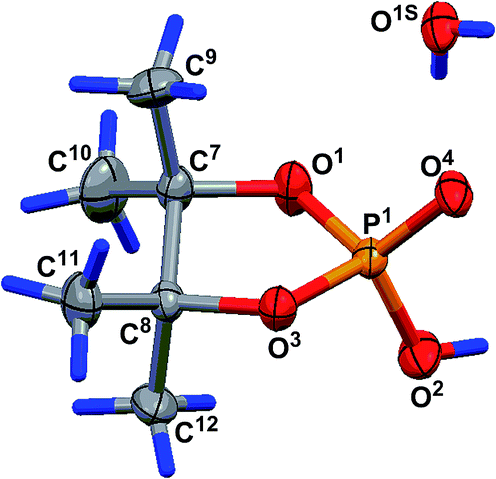 | ||
| Fig. 4 Geometry of molecule 15 in a crystal (solvate with water). Selected bond lengths (Å) and bond and torsion angles (°): P1–O1 1.568(6), P1–O2 1.533(7), P1–O3 1.572(6), P1–O4 1.477(6), O1–P1–O3 98.6(2), O1–P1–O2 108.5(3), O2–P1–O4 113.5(3), O1–C7–C8–O3 36.9(6), C9–C7–C8–C11 33.6(8) and C10–C7–C8–C12 34.5(8). | ||
 | ||
| Fig. 5 System of the intermolecular hydrogen bonds in a crystal of compound 15. | ||
Mild stepwise hydrolysis of P–O-isomer 14 leads to the initial formation of dioxaphospholane 16. The 31P NMR spectrum of this compound contains a doublet (δP 11.0 ppm, 3JPCCH 5.9 Hz) with a coupling constant from the proton at C3, which indicates the retention of the P–O–C3 fragment. Its 13C NMR spectrum shows spin–spin coupling of all the methyl carbons with the phosphorus atom, which clearly points to the retention of the 4,4,5,5-tetramethyl-1,3,2-dioxaphospholane cycle. This data are in accordance with the breaking of the P1–O2 and P1–O7 bonds in phosphorane 14 during hydrolysis. The result of the hydrolysis of phosphorane 14 differs by one for compound 11, which contains a 4,5-dimethyl-1,3,2-dioxaphospholane moiety. In the last case, the 2,3-butanediol elimination proceeds.37
The structure of phospholane 16 was confirmed using single crystal XRD. In Fig. 6, the geometry of the molecule in a crystal is presented (the main geometrical parameters, including bond length and valence and torsion angles of molecule 16 are presented in the ESI†). The phosphorus atom has a distorted tetrahedral configuration. The dioxaphospholane cycle has an envelope conformation with a planar four-membered O1P1O3C7 fragment within 0.080(6) Å, and the C8 atom deviates from this plane by 0.510(9) Å (see Fig. 7). The O2, C10 and C12 atoms are in axial positions (they deviate from the O1P1O3C8 plane by the values of 1.260(5), −1.52(1) and 2.05(1) Å, respectively). The O4, C9 and C11 atoms are in equatorial positions (they deviate from the O1P1O3C8 plane by the values of −1.265(6), 0.74(1) and −0.05(1) Å, respectively). The presence of four methyl groups leads to a noticeable deviation of conformation along the C7–C8 bond from the regular staggered gauche conformation due to steric repulsion (the torsion angles C9C7C8C12 and C10C7C8C11 are equal to −39(1) and −36(1)°, respectively). The envelope conformation is probably realized in a solution for the investigated compound also, which is confirmed by the non-equivalence of these four methyl groups in NMR 1H and 13C spectra, and also by the different spin–spin coupling constants 3JPCCC, which depend on the P–C–C–C torsion angle values. The same situation is realized for the conformation along the C3–C4 bond (the torsion angles of O2C3C4O7 and C13C3C4C16 are 46.2(9) and −45.2(9)°, respectively). The repulsion between the two phenyl groups probably has less meaning in comparison with the phenyl and hexafluoroisopropylhydroxy substituent repulsion, which leads to the same conformation along the C3–C4 bond (the torsion angle of C5C4C3C13 is −166.7(7)°). Due to the classical hydrogen bond, O6–H6⋯O4, the molecules of 16 are connected in dimers, and thus, a 0D supramolecular structure is realized in the crystal [the parameters of the interaction are O6–H6 0.66(6) Å, H6⋯O4 2.14(6) Å, O6⋯O4 2.76(1) Å, ∠O6–H6⋯O4 155(7)°, and symmetry operation 1 − x, 2 − y, −z]. The packing of the molecules in the crystal of 16 is also stabilized by the intramolecular bifurcate O7–H7⋯F1 and O7–H7⋯O2 classical hydrogen bonds [the parameters are O7–H7 0.87(8) Å, H7⋯F1 2.34(9) Å, O7⋯F1 2.860(9) Å, ∠O7–H7⋯F1 119(8)°; and H7⋯O2 2.20(7) Å, O7⋯O2 2.722(8) Å, ∠O7–H7⋯O2 118(6)° respectively, see Fig. 8].
 | ||
| Fig. 6 Geometry of molecule 16 in a crystal (racemate, CR3CS4-enantiomer is shown). Selected bond lengths (Å) and bond and torsion angles (°): P1–O1 1.582(6), P1–O2 1.581(6), P1–O3 1.583(7), P1–O4 1.460(6), O2–C3 1.478(9), O1–P1–O2 101.2(3), O1–P1–O3 99.6(3), O1–P1–O4 118.9(4), O2–P1–O3 109.6(3), O2–P1–O4 112.7(3), O3–P1–O4 113.4(4), P1–O2–C3–C4 163.2(5), O7–C4–C5–O6 165.2(6) and O2–C3–C4–O7 46.1(8). | ||
 | ||
| Fig. 7 Conformation of 1,3,2-dioxaphospholane cycle along the C7–C8 bond in 16 (acyclic substituent is omitted for clarity). | ||
 | ||
| Fig. 8 The system of classical hydrogen bonds in the crystal of 16. | ||
Prolonged hydrolysis of phosphorane 14 leads to the cleavage of not only the exocyclic P1–O6 and P1–O7 bonds but also the 1,3,2-dioxaphospholane P–O bonds. This process is accompanied by the formation of 2,3-dihydroxy-3-trifluoromethyl-4,4,4-trifluoro-1,2-diphenylbutylphosphate 17 and pinacol. It should be noted that phosphate 17 (δP −1.4 ppm, δF −67.05 and −67.82 ppm (CDCl3/DMSO-d6, 3:1)) exists in equilibrium with the cyclic form 19 (δP 14.7 ppm (CD3CN), δF −68.42 and −69.98 ppm (CDCl3/DMSO-d6, 3:1)) (Scheme 5) in a solution.37 The ratio of cyclic and acyclic phosphates in the reaction medium is close to 1:10; however, this equilibrium shifts to the formation of the cyclic derivative 19 upon heating (40 °C) and the ratio becomes 2:1. Long crystallization of the hydrolysis products from CH2Cl2 allowed the isolation of the crystalline complex 18 of the acyclic monophosphate 17 with pinacol in a 2:1 ratio. The structure of complex 18 was confirmed by XRD. The geometry of the complex in a crystal and atom numbering are shown in Fig. 9, and the main geometrical parameters (bond lengths and bond and torsion angles) are listed in the figure caption. The phosphorus atom has a distorted tetrahedral configuration, and the configuration of the chiral atoms is CS3CR4/CR3CS4. The conformations of the monophosphate 17 and pinacol molecules along the C4–C3, C5–C4 and C6–C6a bonds are shown in Fig. 10. All of them are closely related to the almost regular staggered species. The phenyl substituents are located in the gauche-conformation along the C4–C3 bond [the torsion angle of C13–C3–C4–C19 is −50.1(6)°]. The trifluoromethyl groups have different orientations relative to the phenyl ring at the C4 atom (see Fig. 10) and therefore fluorine atoms exhibit non-equivalence in the 19F NMR spectrum owing to the anisotropy of the phenyl ring.
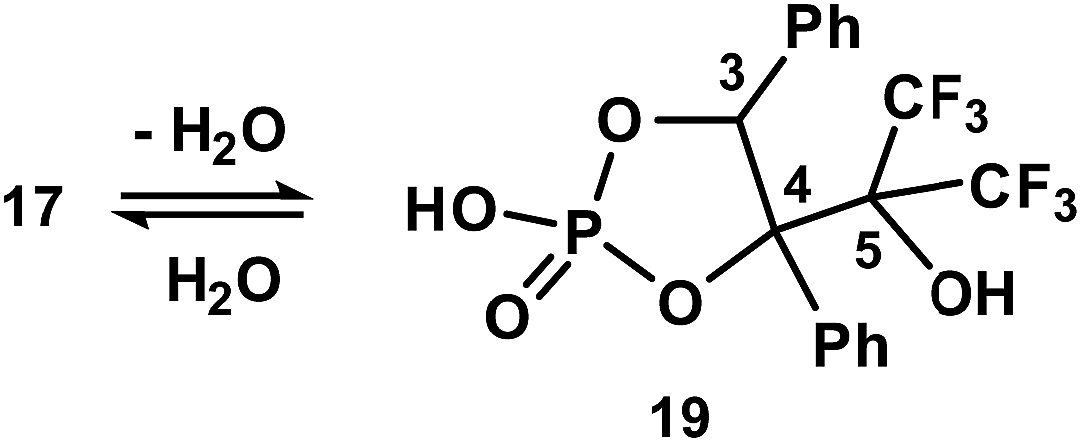 | ||
| Scheme 5 The equilibrium of phosphates 17 and 19 in dioxane solution. | ||
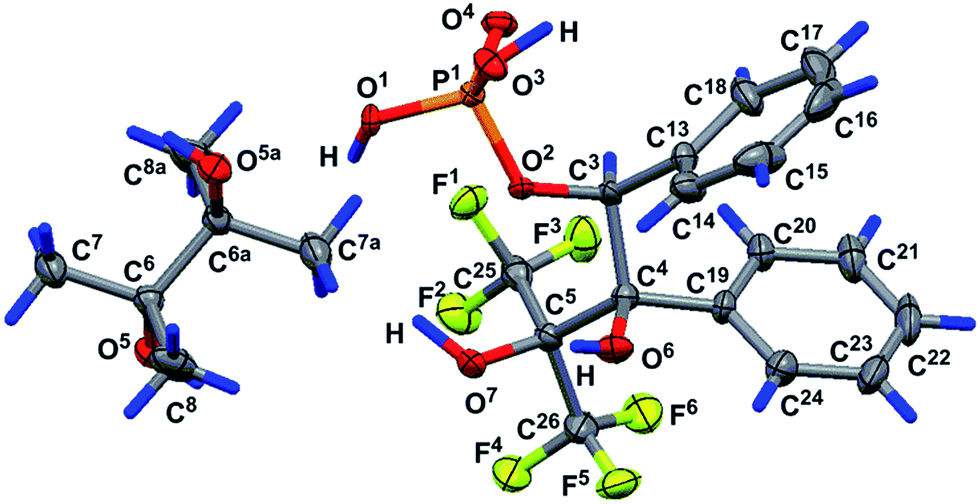 | ||
| Fig. 9 Geometry of complex 18 in a crystal (solvate of molecule 17 with 1/2 pinacol, racemate, and CS3CR4-enantiomer is shown). Selected bond lengths (Å) and bond and torsion angles (°): P1–O1 1.535(5), P1–O2 1.588(4), P1–O3 1.551(4), P1–O4 1.489(4), O1–P1–O2 103.4(2), O1–P1–O3 102.6(3), O1–P1–O4 116.9(3), O2–P1–O3 109.1(2), O2–P1–O4 111.3(2), O2–C3–C4–C5 58.8(6), P1–O2–C3–C4 −168.4(3) and C3–C4–C5–C26 −163.4(5). | ||
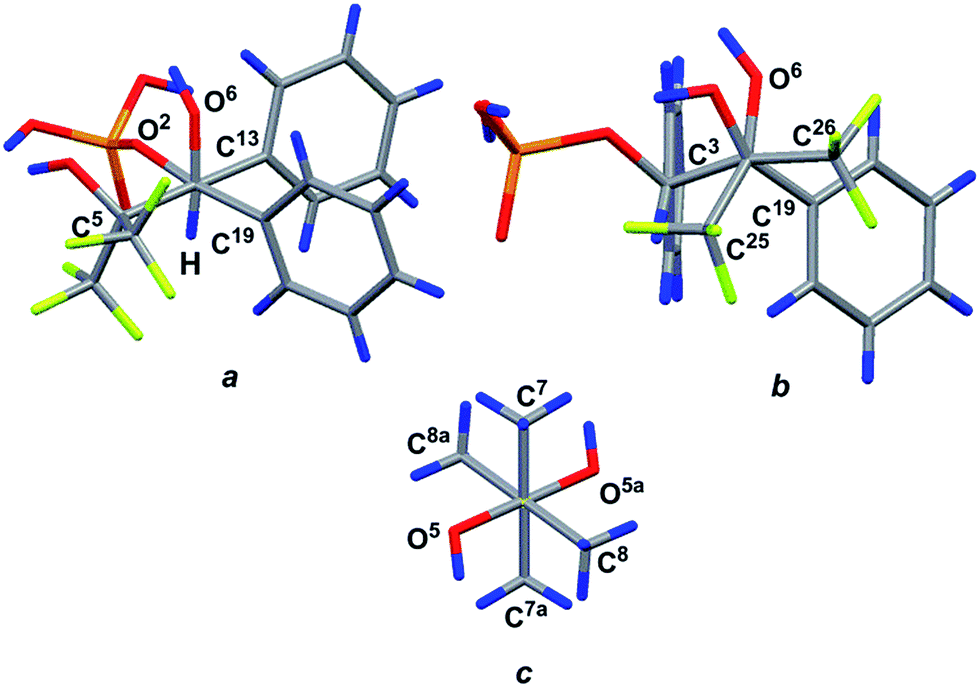 | ||
| Fig. 10 View of the conformations for the molecule (17) in front of the C4–C3 (a), C5–C4 (b) and C6–C6a (c) bonds. | ||
Due to the classical O–H⋯O hydrogen bonds of the molecules, 17 and pinacol form sheets along the b0c crystallographic plane, and thus, a 2D supramolecular structure is realized in the crystal [the parameters of the interactions are O1–H1⋯O(4) interaction: O1–H1 0.86(6) Å, H1⋯O4 1.76(6) Å, O1⋯O4 2.547(6) Å, ∠O1–H1–O4 152(9)°, and symmetry operation x, 3/2 − y, 1/2 + z; O3–H3⋯O5 interaction: O3–H3 0.98(7) Å, H3⋯O5 1.68(7) Å, O3⋯O5 2.639(6) Å, ∠O3–H3–O5 165(5)°, and symmetry operation −1 + x, 1 + y, −1 + z; O7–H7⋯O4 interaction: O7–H7 0.99(9) Å, H7⋯O4 1.85(7) Å, O7⋯O4 2.802(6) Å, ∠O7–H7–O4 159(8)°, and symmetry operation x, 3/2 − y, 1/2 + z]. The packing of the molecules 17 in the crystal of solvate 18 is also stabilized by the intramolecular O7–H7⋯F1 classical hydrogen bond [the parameters are O7–H7 0.99(9) Å, H7⋯F1 2.31(9) Å, O7⋯F1 2.757(6) Å, and ∠O7–H7–F1 106(7)°, see ESI†].
Experimental
1-(2,3-Butylenedioxy)-6,6-bis(trifluoromethyl)-3,4-diphenyl-1,2,5,7-phosphatrioxabicyclo[2.2.11,4]heptane 13
Hexafluoroacetone (4.81 g, 30 mmol) was condensed into a CCl4/CH2Cl2 (1:1) solution (40 mL) of dioxaphosphole 12 (10.4 g, 30 mmol) at −40 °C. The reaction mixture was warmed up to 20 °C (10 h) and then evaporated under reduced pressure (14 Torr) to give a white precipitate of 13, which was filtered off and dried in vacuo (14 Torr). Yield 7.12 g (47%), mp 122–125 °C. The filtrate was evaporated in vacuo (14 Torr) under an argon atmosphere to give a pale yellow oil, which gradually crystallized under a pentane layer (−18 °C). Clear white crystals of compound 13 were filtered off. Yield 3.62 g (24%), mp 123–125 °C. Anal. calcd for C23H23F6O5P (524.12): C, 52.68; H, 4.42; P, 5.91. Found: C, 52.63; H, 4.39; P, 5.98. IR (KBr) (νmax, cm−1): 2987, 2947, 1454, 1398, 1378, 1342, 1277, 1262, 1231, 1199, 1155, 1141, 1080, 1048, 1006, 979, 936, 883, 84, 816, 793, 770, 748, 719, 694, 666, 635, 539, 476. 1H NMR (400 MHz, CDCl3)/δ (ppm): 7.30 (m, 2H), 7.18 (m, 5H), 7.10 (m, 3H), 5.46 (d, 1H, 3JPOCH = 17.0 Hz, H3), 1.49 (br. s, 12H, CH3). 13C/13C–{1H} NMR (100.6 MHz, CDCl3), (hereinafter a view of signal in 13C–{1H} NMR spectrum is in parentheses)/δC (ppm): 135.74 (m (d), 3JHCCC = 7.7 Hz, 2JHCC = 5.2 Hz, 3JPOCC = 0.7 Hz, C13), 133.36 (dt (d), 3JPOCC = 12.2 Hz, 3JHCCC = 7.5 Hz, C19), 129.18 (dt (s), 1JHC = 160.7 Hz, 3JHCCC = 7.4 Hz, C22), 128.61 (dm (s), 1JHC = 161.0 Hz, 3JHCCC = 7.2 Hz, C16), 128.09 (br. dd (s), 1JHC = 160.3 Hz, 3JHCCC = 7.4 Hz, C15,17), 127.90 (dd (s), 1JHC = 161.0 Hz, 3JHCCC = 7.0 Hz, C21,23), 127.21 (br. ddd (s), 1JHC = 159.6 Hz, 3JHCCC = 6.2–6.3 Hz, 3JHCCC = 6.2–6.3 Hz, C14,18), 125.82 (ddd (s), 1JHC = 161.1 Hz, 3JHCCC = 7.4 Hz, 3JHCCC = 6.2 Hz, C20,24), 122.60 (qd (qd), 1JFC = 285.8 Hz, 2JPCC = 1.1 Hz, C26), 121.71 (br. qd (qd), 1JFC = 289.8 Hz, 2JPCC = 5.7 Hz, C25), 99.76 (ddm (d), 2JPOC = 23.9 Hz, 2JHCC = 4.2–4.4 Hz, 3JHCCC = 4.2–4.4 Hz, C4), 87.68 (dmd (d), 1JHC = 156.3 Hz, 2JPOC = 2.9 Hz, 3JHCCC = 4.4 Hz, C3), 82.93 (m (d), 2JPOC = 2.2 Hz, 2JHCC = 4.2–4.4 Hz, Ceq.8), 79.17 (m (d), 2JPOC = 2.9 Hz, 2JHCC = 4.4 Hz, Cax7), 77.78 (d. sept (d. sept), 1JPC = 154.8 Hz, 2JFCC = 30.7 Hz, C6), 23.52 (br. qdq (br. d), 1JHC = 127.3 Hz, 3JPOCC = 11.0 Hz, 3JHCCC = 4.0 Hz, C9–12). 19F NMR (376.5 MHz, CDCl3)/δF (ppm): −68.44 (q. d, 4JFCCCF = 10.3 Hz, 3JPCCF = 4.6 Hz), −68.87 (q, 4JFCCCF = 10.3 Hz, 3JPCCF = 2.3 Hz). 31P–{1H}/31P NMR (161.9 MHz, CDCl3)/δP (ppm): −25.1 (dd (s), 3JPCCH = 17.6 Hz, 3JPCCF = 1.9 Hz). ESI-MS (m/z): 523.97.
1-(2,3-Butylenedioxy)-5,5-bis(trifluoromethyl)-3,4-diphenyl-1,2,6,7-phosphatrioxabicyclo[2.2.11,4]heptane 14
A mixture of compound 13 (6.00 g, 11.45 mmol) and CH2Cl2 (20 mL) was kept for five days (control by 31P NMR). After completion of the rearrangement, the reaction mixture was evaporated in vacuo (14 Torr) under an argon atmosphere to give a yellow oily residue, which was gradually crystallized during storage under a pentane layer (−18 °C). The crystalline precipitate of 14 was filtered off and dried in vacuo (14 Torr). Yield 5.34 g (89%), mp 125–127 °C. Anal. calcd for C23H23F6O5P (524.12): C, 52.68; H, 4.42; P, 5.91. Found: C, 52.64; H, 4.40; P, 5.93. IR (KBr) (νmax, cm−1): 3067, 3036, 2984, 2938, 1500, 1460, 1452, 1396, 1378, 1288, 1241, 1164, 1103, 1042, 1014, 982, 949, 899, 879, 853, 833, 805, 783, 754, 738, 714, 713, 695, 666, 604, 581, 560, 498, 486, 461. 1H NMR (400 MHz, CDCl3)/δ (ppm): 7.44 (m, 1H), 7.30 (m, 1H), 7.17 (m, 3H), 7.08 (m, 5H), 5.72 (d, 1H, 3JPOCH = 19.2 Hz, H3), 1.52 (s, 3H, CH3), 1.51 (s, 3H, CH3), 1.43 (s, 3H, CH3), 1.38 (s, 3H, CH3). 13C/13C–{1H} NMR (100.6 MHz, CDCl3)/δC (ppm): 135.40 (tt (s), 3JHCCC = 5.1–5.3 Hz, 2JHCC = 2.2 Hz, C13), 131.08 (dt (d), 3JPOCC = 18.7 Hz, 3JHCCC = 7.2 Hz, C19), 128.59 (dm (s), 1JHC = 160.7 Hz, 3JHCCC = 7.3 Hz, 3JHCCC = 7.3 Hz, C16 or C22), 128.56 (dm (s), 1JHC = 160.7 Hz, 3JHCCC = 7.3 Hz, 3JHCCC = 7.3 Hz, C16 or C22), 128.52 (dm (s), 1JHC = 161.4 Hz, 3JHCCC = 7.3 Hz, 3JHCCC = 7.3 Hz, C14,18), 127.98 (br. dd (s), 1JHC = 160.5 Hz, 3JHCCC = 7.2 Hz, C23), 127.93 (br. dm (br. sept), 1JHC = 160.5 Hz, 5JFCCCCC = 1.9 Hz, C24), 127.76 (dm (s), 1JHC = 160.7 Hz, 3JHCCC = 6.0–7.0 Hz, C15,17), 127.13 (br. dd (s), 1JHC = 161.4 Hz, 3JHCCC = 7.3 Hz, C21), 126.90 br. dm (br. q), 1JHC = 160.5 Hz, 5JFCCCCC = 5.0 Hz, C20), 122.40 (qd (qd), 1JFC = 286.4 Hz, 3JPOCC = 8.6 Hz, CF3), 121.08 (qd (qd), 1JFC = 289.4 Hz, 3JPOCC = 2.7 Hz, CF3), 83.41 (sept (sept), 2JCCF = 29.3 Hz, C5), 82.93 (m (d), 2JPOC = 2.2 Hz, 2JHCC = 3.6 Hz, 3JHCCC = 3.8 Hz, Ceq.7), 82.47 (dm (dq), 1JHC = 154.8 Hz, 2JPOC = 2.9 Hz, 4JFCCCC = 2.6–2.8 Hz, C3), 79.74 (dt (d), 2JPOC = 20.2 Hz, 3JHCCC = 3.7 Hz, C4), 79.30 (m (d), 2JPOC = 2.9 Hz, 2JHCC = 3.7 Hz, 3JHCCC = 3.6–3.7 Hz, Cax8), 23.72 (qdq (d), 1JHC = 127.6 Hz, 3JPOCC = 6.2 Hz, 3JHCCC = 4.4 Hz, C9–Ceq.7), 23.69 (qdq (d), 1JHC = 127.6 Hz, 3JPOCC = 5.2 Hz, 3JHCCC = 4.4 Hz, C10–Ceq.7), 23.54 (qdq (d), 1JHC = 127.5 Hz, 3JPOCC = 7.7 Hz, 3JHCCC = 4.4 Hz, C11–Cax8), 23.36 (qdq (d), 1JHC = 127.5 Hz, 3JPOCC = 8.0 Hz, 3JHCCC = 4.4 Hz, C12–Cax8). 19F NMR (376.5 MHz, CDCl3)/δF (ppm): −68.22 (q, 4JFCCCF = 9.5 Hz), −72.35 (q, 4JFCCCF = 9.5 Hz). 31P–{1H}/31P NMR (161.9 MHz, CDCl3)/δP (ppm): −27.3 (d (s), 3JPOCH = 20.4 Hz). ESI-MS (m/z): 523.97.Hydrolysis of compound 13
To a suspension of compound 13 (1 g, 1.91 mmol) in diethyl ester (5 mL), a solution of 0.1 mL (5.72 mmol) of water in 3 mL of diethyl ester was added dropwise with constant stirring. The resulting white crystalline precipitate of compound 15 (hydrate with one H2O molecule) was filtered off and dried in vacuo (14 Torr). Yield 0.31 g (83%), mp 190–192 °C. 1H NMR (400 MHz, DMF-d7), δ: 8.72 (br. s, 1H, OH), 1.41 (br. s, 12H, H9–12). 31P–{1H}NMR (161.9 MHz, DMF-d7), δP: 10.6 ppm (s). The 1H NMR spectral data of an aliquot of the filtrate contain the benzoin and hexafluoroisopropanol signals. The structure of hexafluoroisopropanol was proven by a comparison of its spectral characteristics (1H, 13C, 19F NMR) with previously published data.43 The filtrate was evaporated in vacuo (14 Torr) to give a white precipitate of benzoin, mp 132–134 °C. 1H NMR (600.0 MHz, acetone-d6/CDCl3 = 2:1)/δ (ppm): 8.03 (m, 2H, XX′-part of AA′XX′-subsystem, 3JAX = 3JA′X′ = 7.5 Hz, H20,24), 7.56 (tt, 1H, 3JAM = 3JA′M = 7.5 Hz, 4JXM = 4JX′M′ = 1.5 Hz, M-part of AA′MXX′-system, H22), 7.45 (br. dd, 2H, AA′-part of AA′XX′-subsystem, 3JAX = 3JA′X′ = 7.5 Hz, 3JAM = 3JA′M′ = 7.5 Hz, H21,23), 7.44 (m, 2H, XX′-part of AMM′XX′-system, 3JXM = 3JX′M′ = 7.5 Hz, H14,18), 7.32 (br dd, 2H, MM′-part of AMM′XX′-system, 3JXM = 3JX′M′ = 7.5 Hz, 3JAM = 3JAM′ = 7.5 Hz, H15,17), 7.25 (tt, 1H, A-part of AMM′XX′-system, 3JAM = 3JAM′ = 7.5 Hz, 4JAX = 4JAX′ = 1.5 Hz, H16), 6.13 (s, 1H, H3), 4.93 (br. s, 1H, OH). 13C/13C–{1H} NMR (150.0 MHz, acetone-d6/CDCl3 = 2:1)/δC (ppm): 200.01 (br. dt (s), 2JHC3C4 = 3.5 Hz, 3JHC20,24CC4 = 3.3 Hz, C4), 140.75 (tdd (s), 3JHC15,17CC13 = 7.5 Hz, 2JHC3C13 = 5.5 Hz, 3JHOCC13 = 1.3 Hz, C13), 135.36 (br. t (s), 3JHC21,23CC19 = 7.4 Hz, C19), 134.27 (dt (s), 1JHC22 = 162.9 Hz, 3JHC20,24CC22 = 7.7 Hz, C22), 129.86 (ddddd (s), 1JHC20,24 = 161.3 Hz, 3JHC24,20CC20,24 = 7.6 Hz, 3JHC22CC20,24 = 7.6 Hz, 2JHC21,23C20,24 = 1.7 Hz, 4JHC4CCC20,24 = 1.3 Hz, C20,24), 129.52 (br. dm (s), 1JHC15,17 = 161.5–161.8 Hz, 3JHC15,17CC17,15 = 6.0–7.0 Hz, C15,17), 129.41 (dd (s), 1JHC21,23 = 161.7 Hz, 3JHC23,21CC21,23 = 7.6 Hz, C21,23), 128.84 (dtd (s), 1JHC16 = 161.0 Hz, 3JHC14,18CC16 = 7.6 Hz, 5JHC3CCCC16 = 1.1 Hz, C16), 128.44 (dm (s), 1JHC14,18 = 162.4 Hz, 3JHC18,14CC14,18 = 6.5–7.5 Hz, 3JHC16CC14,18 = 6.5–7.5 Hz, 3JHC3CC14,18 = 4.5 Hz, C14,18), 76.94 (dt (s), 1JHC3 = 146.8 Hz, 3JHC14,18CC3 = 4.0 Hz, C3). 13C–{1H} NMR (100.6 MHz, CDCl3), δC: 199.15 (br. dt (s), 2JHC3C4 = 3.3 Hz, 3JHC20,24CC4 = 3.3 Hz, C4), 139.20 (tdd (s), 3JHC15,17CC13 = 7.3 Hz, 2JHC3C13 = 5.5 Hz, 3JHOCC13 = 2.2 Hz, C13), 133.74 (br. t (s), 3JHC21,23CC19 = 7.7 Hz, C19), 134.07 (br. td (s), 1JHC22 = 162.2 Hz, 3JHC20,24CC22 = 7.7 Hz, C22), 129.33 (ddddd (s), 1JHC20,24 = 160.7 Hz, 3JHC24,20CC20,24 = 7.6 Hz, 3JHC22CC20,24 = 7.6 Hz, 2JHC21,23C20,24 = 1.4 Hz, 4JHC4CCC20,24 = 1.2 Hz, C20,24), 129.30 (br. dd (s), 1JHC15,17 = 161.0 Hz, 3JHC15,17CC17,15 = 6.5–7.0 Hz, C15,17), 128.86 (dd (s), 1JHC21,23 = 162.5 Hz, 3JHC23,21CC21,23 = 7.7 Hz, C21,23), 128.76 (dtt (s), 1JHC16 = 161.0 Hz, 3JHC14,18CC16 = 7.2 Hz, 2JHC15,17C16 = 1.5 Hz, C16), 127.96 (dm (s), 1JHC14,18 = 162.4 Hz, 3JHC18,14CC14,18 = 6.5–7.5 Hz, 3JHC16CC14,18 = 6.5–7.5 Hz, 3JHC3CC14,18 = 4.5 Hz, C14,18), 76.42 (dtd (s), 1JHC3 = 146.8 Hz, 3JHC14,18CC3 = 3.4 Hz, 2JHOC3 = 1.5 Hz, C3).
Hydrolysis of compound 14
To a suspension of compound 14 (3.54 g, 6.75 mmol) in 10 mL of diethyl ester, a solution of 0.12 mL (6.75 mmol) of water in 5 mL of diethyl ester was added dropwise with constant stirring. The resulting white crystalline precipitate of compound 16 was filtered off and dried in vacuo (14 Torr). Yield 3.01 g (82%), mp 155–156 °C. Anal. calcd for C23H25F6O6P (542.41): C, 50.93; H, 4.65; P, 5.71. Found: C, 50.94; H, 4.60; P, 5.73. IR (nujol) (νmax, cm−1): 3606, 3214, 2993, 1498, 1451, 1397, 1385, 1262, 1225, 1206, 1145, 1134, 1006, 993, 962, 930, 910, 815, 806, 724, 713, 700, 616, 594, 549, 497, 414. 1H NMR (400 MHz, DMF-d7)/δ (ppm): 8.74 (br. s, 1H, OH), 7.60 (d, 2H, 3JHCCH 5.6 Hz, H20,24), 7.23 (m, 2H, 3JHCCH = 6.7–7.7 Hz, 5JPCCCCH = 1.5 Hz, H14,18), 7.11 (m, 3H, H21–23), 7.05 (m, 3H, H15–17), 6.25 (d, 1H, 3JPOCH = 7.6 Hz, H3), 5.98 (s, 1H, OH), 1.49 (s, 3H, Me), 1.42 (s, 6H, Me), 1.26 (s, 3H, Me). 13C/13C–{1H} NMR (100.6 MHz, DMF-d7)/δC (ppm): 139.45 (br. td (br. s), 3JHCCC = 6.2, 2JHCC = 5.2, C13), 138.09 (ddd (s), 3JHCCC = 7.2 Hz, 3JHCCC = 6.7 Hz, 3JHCCC = 3.7 Hz, C19), 130.91 (dddd (s), 1JHC = 163.3 Hz, 3JHCCC = 5.4 Hz, 3JHCCC = 5.2 Hz, 3JHCCC = 5.2 Hz, C14,18), ∼129.2 (v. br. d (v. br. s) C20), 128.53 (dt (s), 1JHC = 160.0 Hz, 3JHCCC = 7.4 Hz, C16), 128.13 (dt (s), 1JHC = 160.0 Hz, 3JHCCC = 7.7 Hz, C22), 127.69 (br. dd (s), 1JHC = 161.8 Hz, 3JHCCC = 5.0–6.0 Hz, C15,17), 127.37 (v. br. d (v. br. s), 1JHC ∼160.0–162.0 Hz, C21,23,24), 124.80 (q (q), 1JFC = 291.6 Hz, CF3), 124.58 (q (q), 1JFC = 291.1 Hz, CF3), 89.33 (m (d), 2JPOC = 1.2 Hz, C7 or C8), 89.23 (m (s), C8 or C7), 83.34 (sept (sept), 2JFCC = 25.6 Hz, C5), 82.89 (br. dm (br. d), 1JHC = 149.5 Hz, 2JPOC = 5.1 Hz, C3), 81.75 (br. d (d), 3JPOCC = 10.0 Hz, C4), 24.76, 24.27, 24.14 and 23.97 ppm (four qdq (four d), 1JHC = 127.5, 127.5, 127.8 and 127.9 Hz, 3JPOCC = 4.4, 7.0, 3.9 and 6.5 Hz, 3JHCCC = 4.3–4.4 Hz, C9–12). 19F NMR (376.4 MHz, DMF-d7)/δF (ppm): −67.43 (q, 4JFCCCF = 11.3 Hz), −68.19 (q, 4JFCCCF = 11.3 Hz). 31P/31P–{1H} NMR (161.9 MHz, DMF-d7)/δP (ppm): 11.0 (d (s), 3JPCCH = 5.9 Hz). 13C/13C–{1H} NMR (100.6 MHz, DMSO-d6)/δC (ppm): 139.45 (br. td (br. s), C13), 137.0 (m (s), C19), 129.63 (dddd (s), 1JHCCC = 160.0 Hz, 3JHCCC = 6.0–7.0 Hz, 3JHCCC = 6.0–7.0 Hz, 3JHCCC = 4.4 Hz, C14,18), ∼129.2 (v. br. d (v. br. s) C20), 127.65 (dt (s), 1JHC = 160.0 Hz, 3JHCCC = 7.3 Hz, C16), 127.28 (dt (s), 1JHC = 160.0 Hz, 3JHCCC = 7.7 Hz, C22), 126.81 (dd (s), 1JHC = 160.0 Hz, 3JHCCC = 6.6 Hz, C15,17), ∼126.9 (v. br. d (v. br. s), 1JHC ∼160.0–162.0 Hz, C21,23,24), 124.68 (q (q), 1JFC = 292.0 Hz, CF3), 123.45 (q (q), 1JFC = 290.5 Hz, CF3), 89.31 (m (s), C7 or C8), 88.18 (m (s), C8 or C7), 82.01 (sept (sept), 2JFCC = 25.6 Hz, C5), 81.29 (br. dm (br. d), 1JHC = 149.0 Hz, 2JPOC = 4.8 Hz, C3), 80.59 (br. d (d), 3JPOCC = 9.9 Hz, C4), 24.24, 23.72, 23.55 and 23.37 (four qdq (four d), 1JHC = 127.6, 127.6, 128.4 and 129.1 Hz, 3JPOCC = 3.7, 7.0, 3.3 and 6.2 Hz, 3JHCCC = 3.0–3.3 Hz, C9–12). 19F NMR (376.4 MHz, DMSO-d6)/δF (ppm): −66.49 (br. q, 4JFCCCF = 10.9 Hz), −68.10 (br. q, 4JFCCCF = 10.9 Hz). 31P/31P–{1H} NMR (161.9 MHz, DMSO-d6)/δP (ppm): 10.7 (d (s), 3JPCCH = 7.8 Hz). MALDI-MS (m/z): 565.03 (M + Na), 580.93 (M + K).Hydrolysis of compound 16
Compound 16 (1.50 g, 2.77 mmol) was refluxed in a solution (15 mL) of dioxane and H2O (2:1) for 20 hours (control by 31P NMR). 31P/31P–{1H} NMR (161.9 MHz, dioxane/δP (ppm): −1.2 (d (s), 3JPCCH = 5.6 Hz), 14.0 (br. s (s)). 19F NMR (376.4 MHz, CDCl3)/δF (ppm): −67.39 (q, 4JFCCCF = 10.9 Hz), −68.65 (q, 4JFCCCF = 10.9 Hz), −68.65 (q, 4JFCCCF = 10.9 Hz), −69.98 (q, 4JFCCCF = 9.5 Hz). After completion of the hydrolysis, the reaction mixture was evaporated in vacuo (14 Torr) to give a yellow oily residue, which was crystallized from CH2Cl2 (10 mL). The crystalline precipitate of 18 (adduct with pinacol) was filtered off and dried in vacuo (14 Torr). Yield 1.01 g (63%), mp 99–103 °C. Anal. calcd for C23H29F6O8P (578.44): C, 47.76; H, 5.05; P, 5.35. Found: C, 47.74; H, 5.00; P, 5.31. IR (nujol) (νmax, cm−1): 3524, 3499, 3282, 3068, 2349, 1736, 1681, 1601, 1497, 1452, 1377, 1277, 1213, 1154, 1111, 1074, 1022, 1001, 967, 935, 761, 727, 711, 699, 624, 612, 541, 523, 494, 463. 1H NMR (400 MHz, CD3CN)/δ (ppm): 7.46 (br. d, 2H, H20,24), 7.21 (d, 3JHCCH = 7.1 Hz, 2H, H14,18), 7.10 (m, 3H, H19,20,21), 7.04 (m, 3H, H15,16,17), 6.09 (d, 3JPOCH = 4.9 Hz, 1H, H3), 1.16 (s, 12H, Me, pinacol). 13C/13C–{1H} NMR (100.6 MHz, CD3CN)/δC (ppm): 137.98 (br. td (s), 2JHCC = 5.5, C19), 137.07 (br. td (br. s), 3JHCCC 7.3 Hz, C13), 131.10 (br. d (s), 1JHCCC = 156.6 Hz, C14,18), ∼127.9 (v. br. d (v. br. s) C20,24), 128.71 (br. dt (s), 1JHC 161.4 Hz, C16), 128.64 (br. dt (s), 1JHC 161.7 Hz, C22), 128.10 (br. dd (s), 1JHC = 158.5 Hz, 3JHCCC = 7.3 Hz, C15,17), 127.85 and 126.74 (two v. br. d (two v. br. s), C20,24 and C21,23), 124.35 (q (q), 1JFC = 289.4 Hz, CF3), 124.18 (q (q), 1JFC = 290.9 Hz, CF3), 90.34 (m (s), pinacol), 83.79 (sept (sept), 2JFCC = 25.7 Hz, C5), 83.12 (d. m (d), 1JHC = 151.5 Hz, 2JPOC = 2.6 Hz, C3), 81.87 (m (d), 3JPOCC = 8.1 Hz, C4), 25.16 (q (s), 1JHC = 125.4, CH3, pinacol). 19F NMR (376.4 MHz, CDCl3/DMSO-d6 (3:1))/δF (ppm): −67.05 (q, 4JFCCCF = 11.2 Hz), −67.82 (q, 4JFCCCF = 10.2 Hz). 31P/31P–{1H}NMR (161.9 MHz, CD3CN)/δP (ppm): −1.4 (br. s (s)).
Crystal structure determination
Crystal structures were determined by X-ray diffraction of suitable monocrystals. Crystal data were collected at 296 K using graphite monochromatic MoKα (0.71073 Å) radiation and ω-scan. Data collection images were indexed, integrated, and scaled using the APEX2 data reduction package.46 Multi-scan empirical absorption corrections were applied to all data sets, where appropriate, using the SADABS program.47 The structures were solved and refined using the SHELX48 program. All crystal structure pictures were created using Mercury CSD 2.4.49Crystal data for 13
C23H23F6O5P, M = 524.38, monoclinic, a = 9.485(6), b = 11.002(7), c = 12.115(8) Å, β = 101.517(8)°, V = 1239(1) Å3, T = 296 K, space group P21 (no. 4), Z = 2, 12827 reflections measured, 4529 unique (Rint = 0.071), which were used in all calculations. Final indices R1 = 0.0667, wR2 = 0.1629 for 2921 reflections with I > 2σ(I); R1 = 0.1100, wR2 = 0.2021 for all data.
Crystal data for 14
C23H23F6O5P, M = 524.38, orthorhombic, a = 12.650(5), b = 16.695(6), c = 22.988(9) Å, V = 4855(3) Å3, T = 296 K, space group Pbca (no. 61), Z = 8, 28292 reflections measured, 4766 unique (Rint = 0.066), which were used in all calculations. Final indices R1 = 0.0520, wR2 = 0.1426 for 2856 reflections with I > 2σ(I); R1 = 0.0985, wR2 = 0.1825 for all data.
Crystal data for 15
C6H15O5P, M = 198.15, orthorhombic, a = 6.39(2), b = 10.62(3), c = 14.67(3) Å, V = 995(4) Å3, T = 296 K, space group P212121 (no. 19), Z = 4, 5025 reflections measured, 1949 unique (Rint = 0.068), which were used in all calculations. Final indices R1 = 0.0499, wR2 = 0.1437 for 1316 reflections with I > 2σ(I); R1 = 0.0907, wR2 = 0.1841 for all data.Crystal data for 16
C23H25F6O6P, M = 542.40, triclinic, a = 10.788(13), b = 10.889(13), c = 12.329(15) Å, α = 70.63(2), β = 71.74(2), γ = 67.88(2)°, V = 1236(3) Å3, T = 296 K, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), Z = 2, 4706 reflections measured, which were used in all calculations. Final indices R1 = 0.0880, wR2 = 0.2734 for 2147 reflections with I > 2σ(I); R1 = 0.1946, wR2 = 0.3522 for all data.
(no. 2), Z = 2, 4706 reflections measured, which were used in all calculations. Final indices R1 = 0.0880, wR2 = 0.2734 for 2147 reflections with I > 2σ(I); R1 = 0.1946, wR2 = 0.3522 for all data.
Crystal data for 18
C17H15F6O6P 0.5C6H14O2, M = 519.34, monoclinic, a = 18.721(8), b = 13.944(6), c = 8.706(4) Å, β = 95.489(8)°, V = 2262(2) Å3, T = 296 K, space group P21/c (no. 14), Z = 4, 11470 reflections measured, 4448 unique (Rint = 0.152), which were used in all calculations. Final indices R1 = 0.0773, wR2 = 0.1632 for 1683 reflections with I > 2σ(I); R1 = 0.2229, wR2 = 0.2372 for all data.
Conclusions
It was shown that the reaction of 4,4,5,5-tetramethyl-2-(2-oxo-1,2-diphenyl)ethoxy-1,3,2-dioxaphospholane 12 with hexafluoroacetone results in the simultaneous formation of regioisomeric cage (P–C/P–O)-phosphoranes 13 and 14 in the ratio of 10:1. In dichloromethane solution (20 °C, 5 days), the rearrangement of P–C-isomer 13 to P–O-isomer 14 proceeds with high stereoselectivity (>96%). The P–C-isomer hydrolysis unexpectedly leads to the formation of 2-hydroxy-4,4,5,5-tetramethyl-2-oxo-1,3,2-dioxaphospholane with high chemoselectivity. Hydrolysis of the P–O-isomer results in the formation of one stereoisomer of 2-(2,3-dihydroxy-1,2-diphenyl-3-trifluoromethyl-4,4,4-trifluorobutyloxy)-4,4,5,5-tetramethyl-2-oxo-1,3,2-dioxaphospholane with the same configurations for the C3 and C4 atoms, and further hydrolysis of this compound yields 2,3-dihydroxy-3-trifluoromethyl-4,4,4-trifluoro-1,2-diphenyl butylphosphate 17 and pinacol.
Acknowledgements
Financial support from the Russian Foundation for Basic Research (Grant No. 16-03-00451) is gratefully acknowledged.Notes and references
- K. N. Allen and D. Dunaway-Mariano, Trends Biochem. Sci., 2004, 29, 495 CrossRef CAS PubMed.
- L. W. Tremblay, G. Zhang, J. Dai, D. Dunaway-Mariano and K. N. Allen, J. Am. Chem. Soc., 2005, 127, 5298 CrossRef CAS PubMed.
- A. C. Hengge, Adv. Phys. Org. Chem., 2005, 40, 49 CrossRef CAS.
- J. K. Lassila, J. G. Zalatan and D. Herschlag, Annu. Rev. Biochem., 2011, 80, 669 CrossRef CAS PubMed.
- N. J. Baxter, M. W. Bowler, T. Alizadeh, M. J. Cliff, A. M. Hounslow, B. Wu, D. B. Berkowitz, N. H. Williams, G. M. Blackburn and J. P. Waltho, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4555 CrossRef CAS PubMed.
- H. Gu, S. Zhang, K.-Y. Wong, B. K. Radak, T. Dissanayake, D. L. Kellerman, Q. Dai, M. Miyagi, V. E. Anderson, D. M. York, J. A. Piccirilli and M. E. Harris, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 13002 CrossRef CAS PubMed.
- N. J. DeYonker and C. E. Webster, Biochemistry, 2015, 54, 4236 CrossRef CAS PubMed.
- G. Baccolini, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 190, 2173 CrossRef CAS.
- M. A. van Bochove, M. Swart and F. M. Bickelhaupt, J. Am. Chem. Soc., 2006, 128, 10738 CrossRef CAS PubMed.
- M. A. van Bochove, M. Swart and F. M. Bickelhaupt, ChemPhysChem, 2007, 8, 2452 CrossRef CAS PubMed.
- S. Dey, Am. J. Org. Chem., 2015, 5, 83 CAS.
- Best Synthetic Methods: Organophosphorus (V) Chemistry, ed. C. Timperley, Elsevier, 2015, ISBN: 9780080982120 Search PubMed.
- W. Phakhodee, S. Wangngae and M. Pattarawarapan, RSC Adv., 2016, 6, 60287 RSC.
- P. A. Byrne and D. G. Gilheany, Chem. Soc. Rev., 2013, 42, 6670 RSC.
- D. J. Carr, J. J. S. Kudavalli, K. S. Dunne, H. Müller-Bunz and D. G. Gilheany, J. Org. Chem., 2013, 78, 10500 CrossRef CAS PubMed.
- E. V. Jennings, K. Nikitin, Y. Ortin and D. G. Gilheany, J. Am. Chem. Soc., 2014, 136, 16217 CrossRef CAS PubMed.
- K. C. Kumara Swamy, N. N. Bhuvan Kumar, E. Balaraman and K. V. P. Pavan Kumar, Chem. Rev., 2009, 109, 2551 CrossRef PubMed.
- (a) S. Fletcher, Org. Chem. Front., 2015, 2, 739 RSC; (b) D. Camp, M. von Itzstein and I. D. Jenkins, Tetrahedron, 2015, 71, 4946 CrossRef CAS.
- S. Wangngae, C. Duangkamol, M. Pattarawarapan and W. Phakhodee, RSC Adv., 2015, 5, 25789 RSC.
- S. Cao, P. Gao, Y. Guo, H. Zhao, J. Wang, Y. Liu and Y. Zhao, J. Org. Chem., 2013, 78, 11283 CrossRef CAS PubMed.
- W. Dai, Q. Liu, X. You, Z. Zhou, Y. Guo, Y. Zhao and S. Cao, Heteroat. Chem., 2016, 27, 63 CrossRef CAS.
- X. You, L. Qi, J. Zheng, W. Dai, Y. Guo, Y. Zhao and S. Cao, Heteroat. Chem., 2015, 26, 168 CrossRef CAS.
- J. Bader, N. Ignat'ev and B. Hoge, Inorg. Chem., 2014, 53, 7547 CrossRef CAS PubMed.
- S. M. McCarthy, Y.-C. Lin, D. Devarajan, J. W. Chang, H. P. Yennawar, R. M. Rioux, D. H. Ess and A. T. Radosevich, J. Am. Chem. Soc., 2014, 136, 4640 CrossRef CAS PubMed.
- O. Fliss, A. Mejri, K. Essalah, M.-T. Boisdon and B. Tangour, C. R. Chim., 2016, 19, 585 CrossRef CAS.
- N. V. Ignat'ev, J. Bader, K. Koppe, B. Hoge and H. Willner, J. Fluorine Chem., 2015, 171, 36 CrossRef.
- R. Pajkert and G.-V. Röschenthaler, Pentacoordinated and hexacoordinated compounds. Ch. 8, Organophosphorus Chem., 2015, 378–396 CAS.
- (a) R. R. Holmes, Pentacoordinated Phosphorus – Structure and Spectroscopy (ACS Monograph 175), Am. Chem. Soc., Washington, DC, 1980 Search PubMed; (b) R. R. Holmes, Pentacoordinated Phosphorus – Reaction Mechanisms (ACS Monograph 176), Am. Chem. Soc., Washington, DC, 1980 Search PubMed.
- D. E. C. Corbridge, Phosphorus 2000. Chemistry, Biochemistry and Technology, Elsevier, Amsterdam, Lausanne, New-York, Oxford, Shannon, Singapore, Tokyo, 2000, ISBN: 0444825509 Search PubMed.
- L. M. Abdrakhmanova, V. F. Mironov, T. A. Baronova, M. N. Dimukhametov, D. B. Krivolapov, I. A. Litvinov, A. A. Balandina, S. K. Latypov and A. I. Konovalov, Mendeleev Commun., 2006, 16, 320 CrossRef.
- L. M. Abdrakhmanova, V. F. Mironov, T. P. Gryaznova, S. A. Katsyuba and M. N. Dimukhametov, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 652 CrossRef CAS.
- V. F. Mironov, M. N. Dimukhametov, E. V. Mironova, D. B. Krivolapov and L. M. Abdrakhmanova, Russ. J. Gen. Chem., 2015, 85, 441 CrossRef CAS.
- V. F. Mironov, M. N. Dimukhametov, E. V. Mironova, D. B. Krivolapov, G. A. Ivkova and L. M. Abdrakhmanova, Russ. J. Gen. Chem., 2015, 85, 450 CrossRef CAS.
- V. F. Mironov, T. A. Baronova, E. V. Mironova, M. N. Dimukhametov, D. B. Krivolapov and L. M. Abdrakhmanova, Russ. J. Org. Chem., 2015, 51, 401 CrossRef CAS.
- A. N. Pudovik, I. V. Konovalova and E. A. Ishmaeva, Reactions and methods of investigation of organic compounds (In Russ.), ed. B. A. Kazansky, I. L. Knunyants, M. M. Shemyakin and N. N. Melnikov, Chemistry Publish., Moscow, 1973, vol. 23, p. 488 Search PubMed.
- K. Pallitsch, A. Roller and F. Hammerschmidt, Chem.–Eur. J., 2015, 21, 10200 CrossRef CAS PubMed.
- V. F. Mironov, M. N. Dimukhametov, S. V. Efimov, R. M. Aminova, F. K. Karataeva, D. B. Krivolapov, E. V. Mironova and V. V. Klochkov, J. Org. Chem., 2016, 81, 5837 CrossRef CAS PubMed.
- (a) G. V. Romanov, M. S. Yagfarov, A. I. Konovalov, A. N. Pudovik, I. V. Konovalova and T. N. Yusupova, Zh. Obshch. Khim., 1973, 43, 2378 CAS; (b) A. N. Pudovik, I. V. Konovalova, N. P. Anoshina and G. V. Romanov, Zh. Obshch. Khim., 1973, 43, 2153 CAS; (c) E. N. Ofitserov, V. F. Mironov, T. N. Sinyashina, A. N. Chernov, I. V. Konovalova, A. V. Il'yasov and A. N. Pudovik, Dokl. Chem., 1989, 306, 146 Search PubMed.
- N. R. Khasiyatullina, V. F. Mironov, E. V. Mironova, D. B. Krivolapov and I. A. Litvinov, Russ. J. Gen. Chem., 2016, 86, 551 CrossRef CAS.
- J. Pesch, K. Harms and T. Bach, Eur. J. Org. Chem., 2004, 2025 CrossRef CAS.
- S. E. Denmark and Y. Fan, J. Org. Chem., 2005, 70, 9667 CrossRef CAS PubMed.
- M. Baranac-Stojanovic, R. Markovic and M. Stojanovic, Tetrahedron, 2011, 67, 8000 CrossRef CAS.
- T. Hayasaka, Y. Katsuhara, T. Kume and T. Yamazaki, Tetrahedron, 2011, 67, 2215 CrossRef CAS.
- J. R. Cox and M. G. Newton, J. Org. Chem., 1969, 34, 2600 CrossRef CAS.
- L. Labaudiniere, Y. Leroux and R. Burgada, J. Org. Chem., 1987, 52, 157 CrossRef CAS.
- APEX2 (Version 2.1), SAINTPlus. Data Reduction and Correction Program, Version 7.31A, Bruker Advansed X-ray Solutions, BrukerAXS Inc., Madison, Wisconsin, USA, 2006 Search PubMed.
- G. M. Sheldrick, SADABS, Program for empirical X-ray absorption correction, Bruker-Nonis, 1990 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and IR spectra of new compounds and crystallographic data. CCDC 1474601–1474604, 1485824. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra17983e |
| This journal is © The Royal Society of Chemistry 2016 |