
Effervescence-assisted dispersive liquid–liquid microextraction based on the solidification of a floating ionic liquid with a special collection method for the rapid determination of benzoylurea insecticides in water samples
Abstract
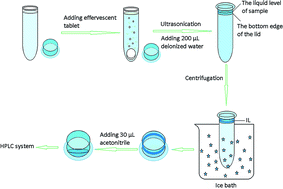
A novel and simple method for effervescence-assisted dispersive liquid–liquid microextraction based on the solidification of a floating ionic liquid using a special collection method combined with high-performance liquid chromatography is developed for the determination of four benzoylureas in water samples. In this method, the ionic liquid (IL), trihexyl(tetradecyl)phosphonium tetrafluoroborate, is used rather than organic drops, which have limitations, as the extraction solvent for the first time in dispersive liquid–liquid microextraction based on the solidification of floating organic drops (DLLME-SFO). Due to the high viscosity of the IL, it can be quickly collected in the lid of the centrifuge tube after solidification, which combines the advantages of both dispersive liquid–liquid microextraction and liquid phase microextraction based on the solidification of a floating IL. Furthermore, effervescence is used to disperse the IL, thereby avoiding the use of an organic dispersant. The influence of various experimental factors, such as the form of the effervescent material, IL quantity, base to acid ratio in the effervescent tablet, weight ratio of IL to effervescent tablet, ultrasonication time, centrifugation time, extraction temperature, sample pH, extraction time and salt addition, are optimized using a central composite design and the one-factor-at-a-time approach. Under the optimal conditions, good linearity was obtained for the four BUs from 2 to 500 μg L−1, with correlation coefficients ranging from 0.9994 to 0.9995. The recoveries were 90.2–100.2% with relative standard deviations ranging from 1.3–4.4%. The limits of detection for the analytes were between 0.77 to 1.58 μg L−1, and the enrichment factors ranged from 206 to 228. Additionally, this method was successfully applied for the determination of four BUs in real water samples.
 Please wait while we load your content...
Please wait while we load your content...

