
Synthesis and characterization of phenol–furfural resins using lignin modified by a low transition temperature mixture
Jun Liu,
Ju Wang,
Yan Fu and
Jie Chang*
School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510640, PR China. E-mail: changjie@scut.edu.cn
First published on 12th September 2016
Abstract
A new modification method based on an oxalic acid/choline low transition temperature mixture was used to activate the chemical groups of lignin as a substitute for phenol in phenol–furfural resins. The optimum modification conditions were 100 °C and 6 h at a molar ratio of oxalic acid to choline chloride of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. The content of phenolic –OH in the modified lignin increased from 1.75 to 3.01 mmol g−1 and the content of –OCH3 decreased from 10.86 to 8.57 wt%, increasing the reactivity of lignin. The modified lignin was partially substituted for phenol at various substitution rates in the synthesis of phenol–furfural (PFU) resins. When the substitution rate of lignin to phenol was 50%, the lignin–PFU had a high bond strength (up to 1.84 MPa) and a low free phenol content and the lignin–PFU showed a higher curing rate and thermostability than PFU alone. The oxalic acid/choline chloride low transition temperature mixture was shown to be an efficient and green lignin modification method for the synthesis of PFU.
:2. The content of phenolic –OH in the modified lignin increased from 1.75 to 3.01 mmol g−1 and the content of –OCH3 decreased from 10.86 to 8.57 wt%, increasing the reactivity of lignin. The modified lignin was partially substituted for phenol at various substitution rates in the synthesis of phenol–furfural (PFU) resins. When the substitution rate of lignin to phenol was 50%, the lignin–PFU had a high bond strength (up to 1.84 MPa) and a low free phenol content and the lignin–PFU showed a higher curing rate and thermostability than PFU alone. The oxalic acid/choline chloride low transition temperature mixture was shown to be an efficient and green lignin modification method for the synthesis of PFU.
1. Introduction
Lignin is a renewable biopolymer with a rich content of aromatic compounds. However, about 5 × 1010 kg of lignin are discharged or burned each year as a by-product of the pulp and paper industries. Lignin is underused because its chemical structure is relatively unreactive.1–5 It would therefore be useful to modify lignin and increase its use in industrial processes.Lignin can be used to replace phenol in the synthesis of phenol–formaldehyde resins because of the large amount of phenolic hydroxyl groups in its structure.6–8 The steric hindrance of lignin is high, however, and it has a high methoxy content, which restricts its use and reduces its practical value.9,10
Common lignin modification methods include sulfonation, graft copolymerization, cross-linking between lignin and acrylamide, acrylic acid and polyhydric monomer ammoxidation, and oxidation in dilute nitric acid.11–16 These methods have a number of shortcomings, such as a high energy consumption and environmental pollution.17,18
The development of deep eutectic solvents (DESs) by Abbott et al.19 provides a new method for the modification of lignin. A DES is composed of two or three components able to self-associate via hydrogen bonding to form a mixture with a melting point lower than that of each individual component.20–23 DESs are referred to as ionic liquid analogues because they have similar physicochemical properties to ionic liquids.24 DESs exhibit impressive properties, such as environmental friendliness, renewability, biodegradability, hypotoxicity and a low production cost. These unique properties have made DESs an important research topic in many different fields.25–27 However, some DESs do not have a eutectic melting point, but instead show a glass transition temperature; these are referred to as low transition temperature mixtures (LTTMs).28
LTTMs or DESs have had an important role in terms of biomass in recent years. LTTMs synthesized from lactic acid and choline chloride have been used as solvents to selectively separate lignin in biomass processing.29,30 A formic acid/choline chloride DES has been shown to increase the accessibility of cellulose by the removal of hemicellulose and lignin.31
Some DESs offer a new method of modifying lignin for phenol–formaldehyde resins. It has been suggested that the lignin processed in a Zn-based DES has an enhanced thermal stability and forms integrated compounds that are useful as fillers for phenol–formaldehyde resins.32 There have been few reports on the activation of the chemical groups of lignin in these ionic liquid analogues. Furfural is regarded as a greener chemical than formaldehyde because it is produced from pentose-based hemicelluloses, another natural resource from biomass.33 It has been considered to be an ideal material in the phenolic resin industry.34
The main goal of the research reported here was to synthesize phenol–furfural (PFU) resins with lignin modified by an oxalic acid/choline chloride LTTM partly replacing the phenol. By determining the effects of different percentages of substitution of lignin on PFU resins, the appropriate amount of modified lignin was determined.
2. Experimental
2.1. Materials
Enzymatic hydrolysis lignin (EHL), furfural (AR, 99%), oxalic acid (AR, 99%), choline chloride (AR, 98.5%), phenol (AR, 99%), sodium thiosulfate (AR, 99%), hydrochloric acid (AR, 99%), potassium bromide (AR, 99%), foline phenol (AR, 99%), urea (AR, 99%), potassium bromate (AR, 99%), hydroxylamine hydrochloride (AR, 99%), vanillin (HPLC, >98%), sodium hydroxide (AR, 99%) and ammonium chloride (99%) were all purchased from Sigma-Aldrich.2.2. LTTM preparation
Oxalic acid (OA) and choline chloride (ChCl) were mixed in specified molar ratios (2:1, 1:1, 1:2, 1:3 and 1:4) and heated in an oil-bath at 80 °C until a transparent homogenous liquid was obtained.
2.3. Modification of lignin in LTTMs
EHL (100 g) was slowly added to 1000 g of each prepared LTTM, then placed into a homogeneous reactor (100 rev/min) at 100 °C for 4 h to determine the optimum molar ratio of OA to ChCl.EHL (100 g) was slowly added to 1000 g of the determined LTTM and placed into a homogeneous reactor (100 rev/min) at 100 °C for different times (2, 4, 6, 8 and 10 h) to determine the optimum reaction time.
EHL (100 g) was slowly added to 1000 g of the determined LTTM and placed into a homogeneous reactor (100 rev/min) at different temperatures (80, 90, 100, 110 and 120 °C) for the optimum time to determine the optimum temperature.
After pre-treatment using these optimum reaction conditions, lignin was recovered by the addition of a hydrochloric acid solution (mass fraction 6%), followed by centrifugation, washing and filtering. The modified lignin (ML) was then dried in a freeze-dryer and the remaining liquid was recovered by vacuum distillation for reuse.
2.4. Characterization of lignin
The methoxy (–OCH3) contents of EHL and ML were determined using the method of Vieböck and Schwappach.35The phenolic hydroxyl (p–OH) contents of EHL and ML were determined by the Folin–Ciocalteau (F–C) assay.36 The F–C analysis was carried out for lignin samples (0.2 g in 50 mL NaOH solution) and the results are expressed as the p–OH content using vanillin as a reference. A calibration graph for vanillin was constructed with seven different concentrations in deionized water (6.000, 12.000, 24.000, 48.000, 60.000 and 84.000 μmol L−1). For the analysis, a 50 μL sample was mixed with 0.6 mL of F–C reagent and 1 mL of Na2CO3 (mass fraction 20%) and diluted to 10 mL with distilled water. The samples were stored in a thermostatic bath at 40 °C for 20 min before measuring their absorbance at 760 nm against a blank (the same reagents free from vanillin). The absorbance (A) at 760 nm of vanillin and the F–C reaction products are shown in Table 1. The relationship between the concentration (c) of vanillin and A (R2 = 0.994) was as follows:
| A = 0.0131c + 0.0507 | (1) |
| c (μmol L−1) | 0 | 6.001 | 12.002 | 24.004 | 48.008 | 61.010 | 84.014 |
| A | 0 | 0.125 | 0.226 | 0.405 | 0.690 | 0.864 | 1.116 |
The p–OH content in the lignin samples was calculated from this equation.
Mw and Mn were determined by gel-permeation chromatography with a UV detector at 240 nm.37 The columns are two cascaded PL-gel columns (5 μm, Agilent Technologies) calibrated with PL polystyrene standards. Acetylated lignin (4 mg) was dissolved in 2 mL of tetrahydrofuran and then 20 μL of the lignin solutions were injected. The column was operated with tetrahydrofuran as the mobile phase at a rate of 1.0 mL min−1 at room temperature.
2.5. Synthesis of PFU and lignin PFU resins
The PFU resins were prepared in a three-necked, round-bottomed flask equipped with a mechanical stirrer, thermometer, cooling condenser and immersed in an oil-bath. The phenol/furfural molar ratio (P/F) was 1:0.8. Phenol (25 g) and NaOH (0.5 g) were charged into the flask and the flask was heated to 45 °C; 23 g of furfural and 0.2 g of urea were then added dropwise. The mixture was heated for 120 min at 135 °C. After the reaction was complete, the flask was cooled to room temperature to stop the reaction. The preparation of the lignin PFU resins followed the same procedure. The substitution rate of lignin to phenol was from 0 to 80%.
2.6. Characterization of PFU and lignin PFU resins
The bond strength was measured as described in ISO 4587-1979. The solids content was measured as described in ISO 760-1978, the viscosity was measured as described in ISO 3219-1993, the free phenol content determined using ISO 8974-2002 and the free aldehyde content was determined using ISO 9397-1995. The structures of PFU and lignin PFU were determined by FTIR spectrometry. The FTIR spectra of the lignin samples were obtained using a Nicolet Tensor 270 spectrometer in the wavenumber range 400–4000 cm−1. Their thermal properties were studied using an STA449C DSC thermal analyser (Netzsch). PFU and MPFU were analysed at 10 °C min−1 under a nitrogen flow-rate of 10 mL min−1. Before testing, the samples were dried for 24 h in a vacuum drying oven to remove any water present. They were then mixed with a suitable amount of urotropine and characterized to compare the curing processes.3. Results and discussion
3.1. Changes in lignin after modification
The –OCH3 content and p–OH content of EHL were 10.86 wt% and 1.75 mmol g−1, respectively.The experiments investigated the effects of the molar ratio of OA to ChCl, the reaction temperature and the reaction time on the changes to the –OCH3 and p–OH contents of the ML.
Fig. 1 shows the effect of the molar ratio of OA to ChCl on the changes in regenerated lignin after reaction at 100 °C for 4 h. OA/ChCl removed –OCH3 and helped to increase the p–OH content at molar ratios of 2:1 to 1:4. When the molar ratio was in the range 2:1 to 1:2, the –OCH3 content decreased while the p–OH content increased, suggesting that the activity of ML was enhanced. This could be attributed to the decrease in the viscosity of LTTM with increasing amounts of ChCl. However, the opposite trend was shown from 1:2 to 1:4. It has previously been reported that hydrogen bond donors, such as OA, can promote the formation of oxygen ions in reactants.38 When the molar fraction of OA decreased, the ability to form oxygen ions was reduced, which meant that it was difficult to form p–OH. Therefore the appropriate molar ratio of OA to ChCl was 1:2 when the –OCH3 content and p–OH content of ML were 9.44 wt% and 2.62 mmol g−1, respectively.
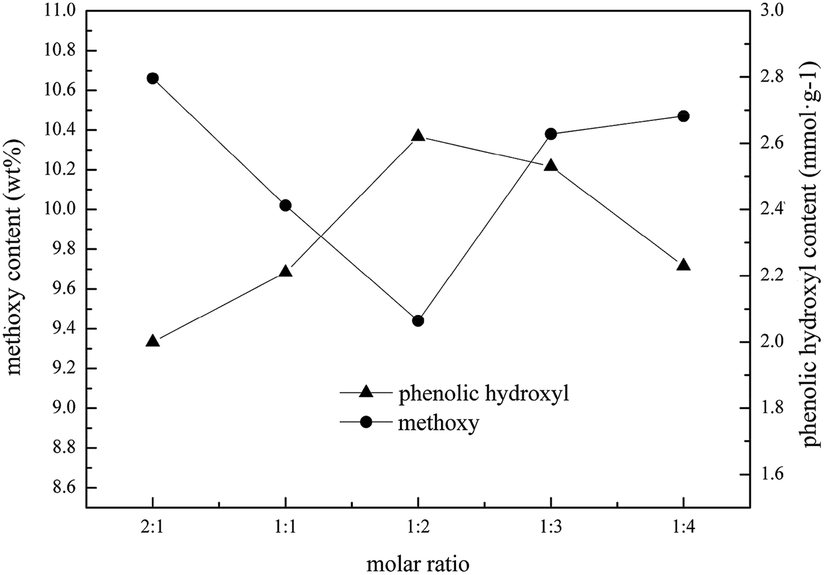 | ||
| Fig. 1 Effect of the molar ratio of OA to ChCl on the changes in lignin. | ||
Fig. 2 shows the effect of the reaction time on the changes to lignin at 100 °C with a molar ratio of 1:2. It could be seen that the content of p–OH increased from 1.75 to 3.01 mmol g−1 in 6 h and then decreased to 2.64 mmol g−1 in the next 4 h. This might be because the degraded lignin underwent a condensation reaction as the time of reaction increased. The –OCH3 content decreased slightly over time because of its extremely stable chemical properties. The optimum reaction time was therefore 6 h.
 | ||
| Fig. 2 Effect of time on the changes in lignin. | ||
Fig. 3 shows the effect of temperature on the changes to lignin at a molar ratio of 1:2 for a reaction time of 6 h. At higher temperatures, more p–OH but less –OCH3 were formed, which was attributed to an increase in the number of ruptured chemical bonds. At temperatures >100 °C, there was little change in the content of either species. This may be because the ability to degrade lignin and the demethoxy in the LTTM had reached saturation point.
 | ||
| Fig. 3 Effect of temperature on changes in lignin. | ||
The best conditions for the modification of lignin were therefore as follows: LTTM molar ratio of OA to ChCl 1:2, reaction time 6 h and temperature 100 °C when the p–OH content of the ML was 3.01 mmol g−1 and the –OCH3 content was 8.57 wt%. The ML used in subsequent work was prepared using these conditions.
Table 2 shows that both the Mw and Mn of ML were significantly lower than in EHL, which suggested that the cross-linked structure of EHL was broken after modification with the DES. The Mw and Mn values of ML were lower than that of EHL, which made ML more active sites exposed, so it benefited the substitution.
| Lignin | Mw | Mn | Polydispersity |
|---|---|---|---|
| EHL | 1755 | 1024 | 1.71 |
| ML | 1353 | 894 | 1.51 |
3.2. Reuse of LTTM
The reuse of the LTTM was studied under the optimum modification conditions for lignin. Fig. 4 shows that both the –OCH3 and the p–OH content of ML changed slightly after four repeated uses, which indicated that the modification effect became poorer and suggested that our recycling rate for LTTM was ideal. | ||
| Fig. 4 Reuse of LTTM. | ||
3.3. Mechanism of lignin modification by the LTTM
The p–OH content increased while the –OCH3 content decreased after LTTM modification and the molecular weight of ML decreased. The chemical structure of lignin may undergo the change shown in Fig. 5 on pre-treatment with the LTTM and precipitation by a hydrochloric acid solution.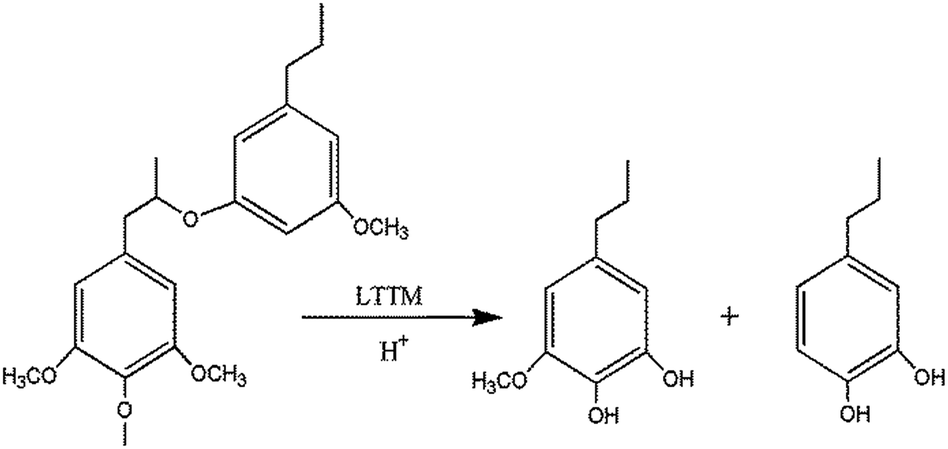 | ||
| Fig. 5 Structural change in lignin modified through the LTTM. | ||
3.4. Effect of rate of substitution of ML to phenol on properties of lignin PFU
Table 3 shows that the bond strength of MPFU was significantly higher than that of EPFU for the same substitution rate of lignin to phenol, which suggested that OA/ChCl had a positive effect on the activation of lignin.| Materials | Bond strength (MPa) |
|---|---|
| PFU with 20% EHL to phenol (EPFU) | 1.07 |
| PFU with 20% ML to phenol (MPFU) | 2.01 |
We investigated the effect of the rate of substitution of ML to phenol on the bond strength and the solid content (Fig. 6). The bond strength of MPFU decreased with an increase in the rate of replacement. However, the decrease in bond strength of MPFU was small when the substitution rate was <50% (from 2.21 to 1.84 MPa) and it decreased sharply when the substitution rate was >50% (from 1.84 to 0.98 MPa). The amount of ML therefore had a negative effect on the bond strength because ML had a lower reactivity than phenol, although it was superior to lignin modified by other methods. The solid content increased significantly (from 41.4 to 46.1%) with an increase in the substitution rate, which favoured a decrease in curing time.
 | ||
| Fig. 6 Changes in solid content and bond strength with rate of substitution. | ||
Fig. 7 shows the effect of the rate of substitution of ML to phenol on free the aldehyde content and free phenol content. The level of free aldehyde increased as a result of the incomplete reaction caused by the increased mass fraction of ML. When the substitution rate was <50%, the free aldehyde content first increased slowly and then sharply. The free phenol content decreased, although the reduction (0.08%) was small between 20 and 80%. The free phenol content changed before and after adding ML. The activity points of ML were less than phenol, which lead to the decrease of molar ratio of p–OH groups to aldehyde groups, making p–OH react more thoroughly.
 | ||
| Fig. 7 Changes in free phenol and aldehyde content with substitution rate of ML to phenol. | ||
Fig. 8 shows the effect of the substitution rate of ML to phenol on the viscosity of MPFU. The viscosity increased with an increasing rate of substitution of ML to phenol. There was a noted increase in viscosity from 50 to 80% because of the exceedingly high content of high molecular weight ML. A high viscosity could lead to an uneven spread of PFU. The curing time of MPFU became shorter as the substitution rate of ML to phenol increased. It has been reported previously that when the concentration and curing temperature were constant, the curing rate of PFU increased as the molar ratio of phenol to furfural decreased.39 The phenol content decreased with an increase in the substitution rate of ML to phenol as a result of the lower content of phenolic hydroxyl groups and active sites of ML, leading to a decrease in the molar ratio of phenol to furfural.
 | ||
| Fig. 8 Changes in curing time and viscosity with substitution rate. | ||
Table 4 shows that when the substitution rate of ML to phenol was 50%, the bond strength of the prepared MPFU was 1.84 MPa, the solid content was 44.1%, the viscosity was 709 mPa s and the free phenol content was 0.31%. All the samples met the Chinese National Standards for adhesives, although the free aldehyde content was 0.39%, a little higher than the Chinese National Standard (Table 4).
| Bond strength (MPa) | Solid content (%) | Viscosity (mPa s) | Free phenol (%) | Free aldehyde (%) | |
|---|---|---|---|---|---|
| CNS | ≥0.7 | ≥35.0 | ≥60.0 | ≤6 | ≤0.3 |
| MPFU | 1.84 | 44.10 | 709 | 0.31 | 0.39 |
Fig. 9 shows the FTIR spectra for PFU and the MPFU resins with 50% substitution of ML to phenol. The 50%-MPFU had a similar FTIR spectrum to PFU, indicating that they had similar chemical structures. The absorption peak at c. 3465 cm−1 in the spectra of phenol–formaldehyde and MPFU was associated with –OH vibrations and there was no distinct difference between the two samples. There were aromatic and furan ring stretching vibration absorption peaks at about 1605, 1515 and 1460 cm−1, also peaks at 835 and 735 cm−1. The absorption peaks at c. 1116 and 1015 cm−1 were associated with C–O–C asymmetrical stretching vibrations. Thus there are many characteristic peaks of the furan ring in the MPFU spectra, indicating that a cross-linking reaction took place between ML and furfural to form macromolecule resins containing furan rings. The strong peak at 1665 cm−1 associated with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in the spectra of MPFU was almost absent in PFU, but strong in MPFU because the free furfural content of MPFU was higher than that of PFU due to the incomplete reaction between ML and furfural.
O in the spectra of MPFU was almost absent in PFU, but strong in MPFU because the free furfural content of MPFU was higher than that of PFU due to the incomplete reaction between ML and furfural.
 | ||
| Fig. 9 FTIR spectra of PFU and 50%-MPFU. | ||
The DSC analyses of PFU and 50%-MPFU were repeated three times; the results are shown in Fig. 10 and Table 5. The onset temperature for 50%-MPFU is 254.6 °C, slightly higher than that of PFU. This suggests that the cross-linking reaction of PFU occurred in a less intensive extension during the preparation because of phenol's more active sites. The curing time of 50%-MPFU was shorter than that of PFU at the same heating rate, which agrees well with the other experimental results. The release of heat from 50%-MPFU was 0.780 w/g, whereas that of PFU was 0.740 w/g, indicating that 50%-MPFU was more thermostable than PFU. This may be a result of the involvement of ML in the synthesis, which led to a significant increase in the apparent Arrhenius activation energy.
 | ||
| Fig. 10 DSC curves for PFU and 50%-MPFU. | ||
| Onset (°C) | Peak (°C) | End (°C) | ΔH (w/g) | |
|---|---|---|---|---|
| PFU-1 | 252.2 | 347.6 | 439.3 | 0.740 |
| PFU-2 | 252.4 | 347.7 | 439.2 | 0.739 |
| PFU-3 | 252.0 | 347.6 | 439.2 | 0.741 |
| PFU-average | 252.2 | 347.6 | 439.2 | 0.740 |
| 50%-MPFU-1 | 254.6 | 349.3 | 437.7 | 0.781 |
| 50%-MPFU-2 | 254.7 | 349.2 | 437.8 | 0.779 |
| 50%-MPFU-3 | 254.6 | 349.1 | 437.8 | 0.780 |
| 50%-MPFU-average | 254.6 | 349.2 | 437.8 | 0.780 |
4. Conclusions
A LTTM based on OA and ChCl offers a new and green approach for the modification of lignin. Lignin modified by an OA/ChCl LTTM showed an obvious improvement in chemical activity and a decrease in molecular weight detected by gel-permeation chromatography.PFU with 50% substitution of ML showed excellent properties and practicality. All the technical parameters met the Chinese National Standards for adhesives, except for the free aldehyde content that was >0.09%.
This work shows that a DES can enhance the chemical activity of lignin and was beneficial as a substitute in phenol–furfural resins. However, the activation mechanism of lignin in LTTM remains obscure, despite the success in modification. Our future research work on OA/ChCl DES will focus on this issue.
Acknowledgements
This work was funded by the National Natural Science Foundation of China (21476090), the National Program on Key Basic Research Project of China (973 Program) (2013CB228104) and the Fundamental Research Funds for the Central Universities.Notes and references
- Y. C. Sun, J. K. Xu, F. Xu, R. C. Sun and G. L. Jones, RSC Adv., 2014, 4, 2743–2755 CAS.
- M. Wu, J. K. Liu, Z. Y. Yan, B. Wang, X. M. Zhang, F. Xu and R. C. Sun, RSC Adv., 2016, 6, 6196–6204 RSC.
- S. S. Y. Tan, D. R. Macfarlane, J. Upfal, L. A. Edye, W. O. S. Doherty, A. F. Patti, J. M. Pringle and J. L. Scott, Green Chem., 2009, 11, 339–345 RSC.
- W. J. Liu, H. Jiang and H. Q. Yu, Green Chem., 2015, 17, 4888–4907 RSC.
- J. W. Jeon, L. Zhang, J. L. Lutkenhaus, D. D. Laskar, J. P. Lemmon, D. Choi, M. L. Nandasiri, A. Hashmi, J. Xu, R. K. Motkuri, C. A. Fernandez, J. Liu, M. P. Tucker, P. B. McGrail, B. Yang and S. K. Nune, ChemSusChem, 2015, 8, 428–432 CrossRef CAS PubMed.
- W. Fang, L. Zhao, F. Liang, H. Chen, S. Gong, Z. Lei and H. Chen, New Carbon Mater., 2015, 95, 1083 CrossRef.
- A. Stücker, F. Schütt, B. Saake and R. Lehnen, Ind. Crops Prod., 2016, 85, 300–308 CrossRef.
- J. Zhou, L. Hu, B. Liang, C. Bo, P. Jia and Y. Zhou, J. Taiwan Inst. Chem. Eng., 2015, 54, 178–182 CrossRef.
- M. V. Alonso, M. Oliet, F. Rodríguez, J. García, M. A. Gilarranz and J. J. Rodríguez, Bioresour. Technol., 2005, 96, 1013–1018 CrossRef CAS PubMed.
- L. Hu, Y. Zhou, R. Liu, M. Zhang and X. Yang, Ind. Crops Prod., 2013, 44, 364–366 CrossRef CAS.
- X. Ouyang, L. Ke, X. Qiu, Y. Guo and Y. Pang, J. Dispersion Sci. Technol., 2009, 30, 1–6 CrossRef CAS.
- M. N. M. Ibrahim, M. R. Ahmed-Haras, C. S. Sipaut, H. Y. Aboul-Enein and A. A. Mohamed, Carbohydr. Polym., 2010, 80, 1102–1110 CrossRef.
- M. N. Satheesh Kumar, A. K. Mohanty, L. Erickson and M. Misra, J. Biobased Mater. Bioenergy, 2009, 3, 1–24 CrossRef.
- M. N. Satheesh Kumar, A. K. Mohanty, L. Erickson and M. Misra, J. Biobased Mater. Bioenergy, 2009, 3, 1–24 CrossRef.
- Q. P. Jiang, Y. X. Zhang, H. T. Mo and Z. H. Li, Environ. Prog., 2006, 25, 251–256 CrossRef CAS.
- J. H. Payne, E. Fukunaga and R. Kojima, J. Am. Chem. Soc., 2002, 7, 1210–1213 Search PubMed.
- M. C. Wang, M. Leitch and C. Xu, Eur. Polym. J., 2009, 45, 3380–3388 CrossRef CAS.
- A. Effendi, H. Gerhauser and A. V. Bridgwater, Renewable Sustainable Energy Rev., 2008, 12, 2092–2116 CrossRef CAS.
- A. P. Abbott, D. Boothby, G. Capper, D. L. Davies and R. K. Rasheed, J. Am. Chem. Soc., 2004, 126, 9142–9147 CrossRef CAS PubMed.
- A. Wang, P. Xing, X. Zheng, H. Cao, G. Yang and X. Zheng, RSC Adv., 2015, 5, 59022–59026 RSC.
- W. Zaidi, L. Timperman and M. Anouti, RSC Adv., 2014, 4, 45647–45652 RSC.
- M. C. Gutiérrez, D. Carriazo, C. O. Ania, J. B. Parra, M. L. Ferrer and F. Monte, Energy Environ. Sci., 2011, 4, 3535–3544 Search PubMed.
- T. J. Trivedi, J. H. Lee, H. J. Lee, Y. K. Jeong and J. W. Choi, Green Chem., 2016, 18, 2834–2842 RSC.
- H. C. Hu, Y. H. Liu, B. L. Li, Z. S. Cui and Z. H. Zhang, RSC Adv., 2015, 5, 7720–7728 RSC.
- Y. Dai, J. Spronsen, G. J. Witkamp, R. Verpoorte and Y. H. Choi, J. Nat. Prod., 2013, 76, 2162–2173 CrossRef CAS PubMed.
- L. Gu, W. Huang, S. Tang, S. Tian and X. Zhang, Chem. Eng. J., 2015, 259, 647–652 CrossRef CAS.
- A. Procentese, E. Johnson, V. Orr, A. G. Campanile, J. A. Wood, A. Marzocchella and L. Rehmann, Bioresour. Technol., 2015, 192, 31–36 CrossRef CAS PubMed.
- M. Francisco, A. S. B. González, S. L. G. D. Dios, W. Weggemans and M. C. Kroon, RSC Adv., 2013, 3, 23553–23561 RSC.
- M. Francisco, A. V. D. Bruinhorst and M. C. Kroon, Angew. Chem., Int. Ed., 2013, 52, 3074–3085 CrossRef CAS PubMed.
- M. Francisco, A. V. D. Bruinhorst and M. C. Kroon, Green Chem., 2012, 14, 2153–2157 RSC.
- G. C. Xu, J. C. Ding, R. Z. Han, J. J. Dong and Y. Ni, Bioresour. Technol., 2016, 203, 364–369 CrossRef CAS PubMed.
- H. Lian, S. Hong, A. Carranza, J. D. Mota-Morales and J. A. Pojman, RSC Adv., 2015, 5, 28778–28785 RSC.
- F. B. Oliveira, C. Gardrat, C. Enjalbal, E. Frollini and A. Castellan, J. Appl. Polym. Sci., 2008, 109, 2291–2303 CrossRef CAS.
- I. Agirrezabaltelleria, C. Garcíasancho, P. Mairelestorres and P. L. Arias, Chin. J. Catal., 2013, 34, 1402–1406 CrossRef CAS.
- F. Vieböck and A. Schwappach, Eur. J. Inorg. Chem., 1930, 63, 2818–2823 Search PubMed.
- A. García, M. G. Alriols, G. Spigno and J. Labidi, Biochem. Eng. J., 2012, 67, 173–185 CrossRef.
- J. L. Wen, S. L. Sun, B. L. Xue and R. C. Sun, J. Agric. Food Chem., 2013, 61, 635–645 CrossRef CAS PubMed.
- H. R. Lobo, B. S. Singh and G. S. Shankarling, Catal. Lett., 2012, 142, 1369–1375 CrossRef CAS.
- Y. Chen, J. Chang and J. Fan, Chem. Ind. Eng. Prog., 2011, 30, 306–312 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |