
Identification of a novel human DNA ligase I inhibitor that promotes cellular apoptosis in DLD-1 cells: an in silico and in vitro mechanistic study†
Deependra Kumar Singha,
Mohd. Kamil Hussain‡
b,
Shagun Krishnaa,
Amit Laxmikant Deshmukha,
Mohammad Shameema,
Pooja Mauryaa,
Kanchan Hajela*bc,
Mohammad Imran Siddiqi*ac and
Dibyendu Banerjee*ac
aMolecular and Structural Biology Division, CSIR-Central Drug Research Institute, Sector 10, Jankipuram Extension, Sitapur Road, Lucknow 226031, UP, India. E-mail: d.banerjee@cdri.res.in
bMedicinal and Process Chemistry Division, CSIR-Central Drug Research Institute, Sector 10, Jankipuram Extension, Sitapur Road, Lucknow 226031, UP, India
cAcademy of Scientific and Innovative Research (AcSIR), India
First published on 26th September 2016
Abstract
The processes of DNA replication and repair are accomplished by the concerted action of several proteins. Among them human DNA ligases play an important role during the last step of almost all DNA replication and repair processes, where they seal the nicks between DNA strands. In humans, three kinds of DNA ligases (human DNA ligase I, III, IV) are found. DNA ligase I (hLigI) is involved in both DNA replication as well as in DNA repair pathways and is reported to be over-expressed in rapidly dividing cells, including cancer cells. For this reason, in this study we have targeted hLigI for studying its response as a novel anticancer target. We have screened for ligase I inhibitors from our in-house small molecule library by a previously validated pharmacophore based virtual screening method and found a novel hLigI inhibitor. This compound (S-097/98) demonstrated antiproliferative activities specifically in DLD-1 (colon), MDA-MB-231 (triple negative breast) and HepG2 (liver) cancer cell lines at low micromolar concentrations of 6–7 μM. Mechanistic studies show that the compound can directly interacts with the hLigI protein and inhibits ligation of both the purified protein in vitro, as well as in cell lysate of DLD-1 cells treated with the inhibitor. The compound also arrests cell cycle progression at the G2/M phase and increases the nuclear size of DLD-1 cancer cells, thereby demonstrating its antiproliferative activity. Finally, the compound promotes cellular apoptosis in DLD-1 cells.
1. Introduction
The successful survival and propagation of living organisms requires an error free and intact transfer of genetic material from one generation to the next.1 The maintenance of genomic integrity in this process is not an easy job considering that vast amounts of DNA in cells are always subject to damage from exogenous as well as endogenous factors. To counter this, cells have developed highly accurate DNA replication and repair pathways that help them maintain their genomic integrity.2,3 There are several DNA repair pathways working inside the cells, namely Fanconi anemia (FA) repair pathway,4 homologous recombination (HR),5,6 non-homologous end joining (NHEJ),7 base excision repair (BER),8 nucleotide excision repair (NER),9 direct repair (DR),10 mismatch repair (MMR),11 translesion synthesis (TLS),12 etc. These repair pathways involve an interplay of various proteins such as apurinic–apyrimidinic exonuclease 1 (APEX1), Ataxia Telangiectasia Mutated (ATM), Ataxia Telangiectasia and Rad3-related (ATR), Cell Division Control 25 (CDC25), cyclin-dependent kinase (CDK), checkpoint kinase (CHK), CDC-like kinase 2 (CLK2), DNA methyltransferase 1 (DNMT1), flap structure-specific endonuclease 1 (FEN1), DNA ligases, O-6-methylguanine-DNA methyltransferase (MGMT), poly(ADP-ribose)glycohydrolase (PARG), poly(ADP-ribose)polymerase (PARP), DNA polymerase (POL), sirtuin 1 (SIRT1), tyrosyl-DNA phosphodiesterase 1 (TDP1), telomere reverse transcriptase (TERT) and topoisomerases (TOP), DNA-dependent protein kinase (DNA-PK).13 Many of these proteins are now being studied for their drugability for different diseases.14 Among these, DNA ligases have recently emerged as potential targets for the development of novel anticancer molecules.15–23DNA ligases are very important enzymes involved in almost all DNA replication and repair processes where their major role is the sealing of single strand (ss) and double strand (ds) breaks between DNA strands. DNA nick sealing is a three step process where, in the first step, ligases are adenylated by using the cofactor ATP (in eubacteria, archaebacteria and eukaryotes) or NAD (in eubacteria). In the second step the adenylated group is transferred to the 5′-phosphate of the nicked DNA molecule, and in the third step, the 3′-OH end of the DNA strand attacks the 5′-PO4 of the adjacent strand to release AMP and seals the nick by making a phosphodiester bond between the ends of the DNA to ligate them.24–26
In humans, three types of DNA ligases (human DNA ligase I, III, IV/XRCC4) are found. Although they are encoded by different genes, they have similar catalytic regions and perform somewhat overlapping functions in DNA replication and repair pathways.17,27–30 Human DNA ligase I (hLigI) is mainly involved in the joining of nascent DNA (Okazaki fragments) generated during the process of replication31 and is also involved in single strand break repair pathways (SSBR) such as long patch base excision repair (BER) pathway32–34 and micro-homology mediated end-joining (MHEJ) pathway which is a form of double strand break repair pathway.35 Human DNA ligase III (hLigIII) is involved in single strand break repair (SSBR) pathways like short patch base excision repair (BER) pathway and nucleotide excision repair (NER) pathway.36 Human DNA ligase IV (hLigIV/XRCC4) performs DNA nick sealing in double strand break repair pathway (DSBR) by non-homologous end joining (NHEJ).37 Looking at the functions performed by DNA ligases, it seems that they play very significant roles in maintaining genomic integrity.
We therefore predicted that inhibiting the function of ligases in cells will lead to difficulty in cell survival due to accumulation of DNA strand breaks and hence DNA ligases have the potential to work as novel anticancer targets. However, ligases are also expressed in normal cells and in theory ligase inhibitors might also affect normal cells. However, the specificity issue can be overcome by targeting the hLigI enzyme which has been reported to be over-expressed in rapidly dividing cells and cancer cells.38,39 Ligase I inhibitors could be used for cancer therapy because on the one hand they can block cellular replication and on the other hand, in combination with DNA damaging agents (such as doxorubicin, cisplatin or radiation therapy), they can also presumably block the DNA repair pathways, so that cancer cells will preferentially undergo programmed cell death or apoptosis.
Although several compounds that can inhibit ligases have been reported, none are currently used in therapy. In a step towards that goal we tested whether ligase inhibitors can show good antiproliferative and anticancer activities. As reported earlier,19 we employed an in silico followed by an in vitro approach where we conducted a pharmacophore based virtual screening of our in-house [Central Drug Research Institute (CDRI)] small molecule library and thereafter the top hits obtained from the screening were tested in our in vitro studies to confirm their antiligase and antiproliferative activities.
2. Experimental section
2.1. Reagents, media and antibodies
DNA oligos were purchased from Integrated DNA technologies (IDT, India). MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide), trypsin–EDTA, propidium iodide, DMSO and protease inhibitor cocktail were purchased from Sigma-Aldrich (St. Louis, MO, USA). Cell growth media DMEM and EMEM were purchased from Sigma-Aldrich (St. Louis, MO, USA), McCoy's 5A media was purchased from Cell clone (Genetix, India), RPMI 1640, FBS and antibiotic and antimycotic solution were purchased from Invitrogen (Whitefield, Bangalore). Fluorescein isothiocyanate (FITC)-labelled Annexin V (Annexin V-FITC) kit were obtained from BD Biosciences (San Diego, CA). Antibodies against human poly(ADP-ribose)polymerase (PARP), γH2AX, cleaved caspase 3 and procaspase 3 were purchased from Cell Signalling Technology (CST, Beverly, MA), USA. Secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), β-actin antibody was purchased from Sigma-Aldrich (St. Louis, MO).2.2. In silico pharmacophore based screening of ligase inhibitors from CDRI compound library
The ligand based virtual screening procedure to identify compounds that may have a likelihood of inhibiting hLigI involved several steps including pharmacophore generation and validation, virtual screening of our Institutional small molecule database (CDRI database) followed by molecular docking studies of the identified hits. The pharmacophore model used in this study was derived using three structurally diverse most active inhibitors of hLigI identified in a study done by Tomkinson et al.15 and described previously.19 The inhibitors were sketched, energy minimization was done and then they were used to yield a set of pharmacophores applying GASP program integrated with Sybyl7.1.40 The pharmacophore models were statistically validated according to various parameters. The most statistically significant pharmacophore Hypo1 comprises of three features as identified by GASP: one hydrophobic feature (HYD) and two acceptor atoms (AA). Details of the pharmacophore generation and validation have been described in our previous study.19,20 In the present study, the statistically most significant pharmacophore model Hypo1 was used for the screening of our in-house small molecule compound database. The database of approximately 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 small molecules has been developed at the CSIR-Central Drug Research Institute (CDRI), Lucknow, India. During preparation of the structures of the compounds in the database, hydrogen atoms were added and geometry optimization was performed with the help of MMFF94 force field using Sybyl7.1. Virtual screening was performed using Unity module of Sybyl7.1.41 For the docking of compounds retrieved through pharmacophore search, crystal structure of hLigI bound to 5′-adenylated, nicked DNA was used.42 Prior to docking, the hLigI structure was prepared by removing deoxyribonucleotide moieties and adding hydrogen atoms. It was further subjected to energy minimization by Sybyl7.1 applying default settings. The active site for docking was selected after thorough visual inspection after considering the DNA binding residues known from PDB ID: 1X9N. Docking was performed using FlexX module embedded with Sybyl7.1.43 The full procedure for selection, validation of active site and docking studies can be obtained from our previous studies.19,20
500 small molecules has been developed at the CSIR-Central Drug Research Institute (CDRI), Lucknow, India. During preparation of the structures of the compounds in the database, hydrogen atoms were added and geometry optimization was performed with the help of MMFF94 force field using Sybyl7.1. Virtual screening was performed using Unity module of Sybyl7.1.41 For the docking of compounds retrieved through pharmacophore search, crystal structure of hLigI bound to 5′-adenylated, nicked DNA was used.42 Prior to docking, the hLigI structure was prepared by removing deoxyribonucleotide moieties and adding hydrogen atoms. It was further subjected to energy minimization by Sybyl7.1 applying default settings. The active site for docking was selected after thorough visual inspection after considering the DNA binding residues known from PDB ID: 1X9N. Docking was performed using FlexX module embedded with Sybyl7.1.43 The full procedure for selection, validation of active site and docking studies can be obtained from our previous studies.19,20
2.3. In vitro DNA ligation assays
DNA ligation assays were performed as described in our previous work.19 We have used three different single strand DNA oligos, 52-mer (5′-GTACGTCGATCGATTGGTAGATCA GTGTCTATGTATGTCAGTGAGATAGTAC-3′), 25-mer (5′-CTGATCTACCAATCGATCG ACGTAC-3′) and 27-mer (5′-/5Cy3/GTACTATCTCACTGACATACATAGACA-3′) and annealed them together to form a double-stranded nicked substrate for the ligase enzymes. For the easy detection of oligos the 5′ end of 27-mer oligo was labelled with a fluorescent dye cyanin 3 (Cy3). The reaction mixture (20 μL) contained 1 pmol of labelled DNA substrate, 0.2 pmol of purified hLigI and inhibitors or solvent control (DMSO) in a ligation buffer containing Tris–Cl (50 mM, pH 7.5), MgCl2 (10 mM), BSA (0.25 mg mL−1), NaCl (100 mM) and ATP (500 μM) at 37 °C for 30 min. Reactions were stopped by adding 10 μL of stop buffer (90% formamide and 10% of 50 mM EDTA). The ligated and unligated DNA molecules were separated in denaturing gel containing 7 M urea and 12% acrylamide. Images were captured and bands were quantified by image quant LAS 4010 (GE Healthcare, Little Chalfont, Buckinghamshire).2.4. Electrophoretic mobility shift assay (EMSA)
EMSA was performed as described previously.19 We have used a non-ligatable, nicked, double stranded DNA substrate. Briefly, the substrate is made up of three oligos, one was a 5′-FAM labelled 27-mer oligo with a dideoxy modified 3′ end (5′-/56-FAM/GTACTATCTCAC TGACATACATAGAC/3ddc/-3′), where FAM was used for easy detection of DNA substrate and dideoxy modification was used for creating the non ligatable nick. The other two oligos were a 25-mer (5′-CTGATCTACCAATCGATCGACGTAC-3′) and a 52-mer (5′-GTACGTCGATCGATTGGTAGATCAGGGTCTATGTATGTCAGTGAGATAGTAC-3′). The oligos were annealed to obtain a non-ligatable nicked substrate for hLigI protein. We incubated 10 pmol of hLigI with 50, 100, 250, 500 μM inhibitor and 2 pmol of DNA substrate in a ligation buffer containing Tris–Cl (50 mM, pH 7.5), MgCl2 (10 mM), BSA (0.25 mg mL−1), NaCl (100 mM) and ATP (500 μM), in a reaction volume of 20 μL, for two hours on ice. After the addition of 10 μL of native gel buffer [50 mM Tris–Cl (pH 7.5), 20% glycerol, 0.05% bromophenol blue], samples were separated by 6.5% native PAGE and bands were detected by Image Quant LAS4010.2.5. DNA intercalation assay
To determine whether the inhibition of ligation occurs due to non-specific DNA intercalation rather than direct protein binding, we performed the DNA intercalation assay with the inhibitor as described previously.19 We incubated different concentrations (100, 250, 500 μM) of S-097/98 with 100 ng of linearized pUC18 plasmid for 30 min at 37 °C in ligation buffer. After the incubation, the reaction products were separated on a 1% agarose gel at 5.3 V cm−1. Gels were visualized by ethidium bromide staining. Here we included doxorubicin (a known DNA intercalator) as positive control and ampicillin as negative control for DNA intercalation assays.2.6. DNA and protein cleavage assay
To check the direct interaction of compound with DNA and protein, we have applied the concept of DARTS (drug affinity responsive target stability).44 To study the interaction between DNA and compound, we incubated 100 ng of DNA (pUC18) with 250 and 500 μM of compound for 30 min at 37 °C in ligation buffer. After incubation, the DNA was cleaved by adding 0.0025 U of DNase1 in each reaction mixture for 5 min at 37 °C and the reaction was stopped by adding stop buffer which contained 7 M urea, 20 mM Tris–Cl (pH 7.5), 50 mM EDTA, 20% glycerol and bromophenol blue.To study the interaction between protein and compound we incubated the purified hLigI protein or DLD-1 cell lysate with 250 and 500 μM concentration of compound in ligation buffer for two hours on ice. After incubation, purified as well as cell lysate proteins were digested by porcine trypsin (1 μg) at 37 °C for 30 min. The protein bands were detected either by silver staining in case of purified protein or by Western blotting in case of cell lysate respectively (Fig. 3B and ESI Fig. 2†).
2.7. Cell culture and viability assays
The hepatic and breast cancer cell lines HepG2 and MDA-MB-231 (ATCC, Manassas, VA) were grown in DMEM. Colon cancer cell line DLD-1 (ECACC, Salisbury, UK) was cultured in RPMI 1640. HEK-293 (ATCC, Manassas, VA) and 46Br.1G1 (ECACC, Salisbury, UK) were cultured in EMEM media. Media were supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% antibiotic–antimycotic solution. Compounds to be screened were obtained as powder from CDRI small molecule library and dissolved in DMSO (Sigma St. Louis, MO) as 10 mM stocks. Cells (5000 per well) to be tested were plated in 96 well plates. After incubating overnight, the cells were treated with five different concentrations of compounds (50, 25, 12.5, 6.25 and 3.12 μM) in triplicates for 48 h. Subsequently, 10 μL of MTT (5 mg mL−1) was added to each well and incubated for 3 h. The MTT formazan formed by viable cells was dissolved in 100 μL of DMSO and shaken for 10 min or until it dissolved. Then absorbance was measured at 590 nm on a plate reader (Epoch Microplate Reader, Biotek, USA). The cytotoxic effects of compounds were calculated as % inhibition in cell growth as per the formula [100 − (absorbance of inhibitor treated cells/absorbance of control cells)] × 100.2.8. Apoptosis and cell cycle distribution analysis
Apoptotic cell death of DLD-1 cells upon treatment with S-097/98 was measured using a commercially available Annexin V-FITC apoptosis detection kit according to the manufacturer's protocol (BD Biosciences, San Diego, CA). Briefly, cells were treated with different concentrations (7, 10 and 14 μM) of S-097/98 for 48 h. The cells were then harvested, washed with cold PBS and stained with Annexin V-FITC and propidium iodide (PI) in binding buffer at room temperature in the dark. The stained cells were analyzed by fluorescence-activated cell sorting using a FACS-Calibur instrument (BD Biosciences, San Diego, CA) equipped with CellQuest 3.3 Software.For the study of cell cycle distribution, cells were synchronized by serum starvation for 36 h and then grown in serum containing media in the presence or absence of 7 μM (IC50 concentration) of S-097/98. After 0, 12 and 24 h, cells were harvested, washed twice with cold PBS and fixed with 70% ethanol. The fixed cells were washed with chilled PBS and then stained with 50 μg mL−1 of propidium iodide. Cell cycle distribution was analyzed by using FACS-Calibur (BD Biosciences, San Diago, CA).
2.9. Estimation of nuclear size
The effect of compound on the size of the nuclei of DLD-1 cells was determined by fluorescence microscopy and flow cytometric pulse width analysis (FL2-W) after cells were stained with propidium iodide.45,46 DLD-1 cells were grown on coverslips and synchronized by serum starvation. After serum starvation cells were treated with 7 μM (IC50 concentration) of compound for 24 h, cells were fixed in 4% paraformaldehyde for 30 min at room temperature and nuclei were stained with propidium iodide (PI). The images were captured by fluorescence microscope (Nikon Eclips Ti-S) and analysed by CellProfiler 2.1.1 image analysis software.2.10. Preparation of cell lysate and Western blotting
Cells (DLD-1) were seeded in 6 well plates and treated with different concentrations (0, 7 and 14 μM) of S-097/98. After 48 h, cells were harvested and washed twice with chilled PBS. Thereafter the cells were suspended in chilled PBS containing 1× protease inhibitor cocktail and lysed by very brief sonication (2 seconds on followed by 10 second off cycle for 3 times) on ice. Sonicated samples were centrifuged at 10000 rpm for 30 min at 4 °C and supernatant was collected and used for ligation assay and for Western blotting. Protein samples were separated by SDS PAGE and proteins were transferred to PVDF membranes. The membranes were probed with the indicated primary antibodies, followed by the appropriate HRP-conjugated secondary antibodies, and developed by enhanced chemiluminescence in Image Quant LAS 4010 (GE Healthcare, Little Chalfont, Buckinghamshire).
2.11. Statistical analysis and IC50 calculation
All experiments were performed in triplicate and the data represented as mean ± SEM. All data was analysed using the Graph Pad Prism software version 5 (San Diego California USA). For the measurements of apoptosis, cell cycle analysis, ligation activity of cell lysate and differential expression of proteins in Western blots, Student's t-test and one-way ANOVA followed by Newman–Keuls test were used to assess the statistical significance between control and treated groups. A statistically significant difference was considered at the level of P ≤ 0.05. With the help of Prism software, IC50 values were calculated by using nonlinear regression option to fit the data to the log(inhibitor) vs. normalized response curve.3. Results and discussion
3.1. In silico pharmacophore based screening of ligase inhibitors from CDRI compound library
As a result of virtual screening, we have identified 7636 compounds that mapped to the pharmacophoric features of the Hypo1. These compounds were further docked inside the active site of hLigI. We have applied FlexX module to predict reasonable binding mode of these compounds. To predict the docking score, we have used the default FlexX parameters. The compounds that docked into hLigI active site were ranked according to the FlexX docking energy. In accordance with our previous study, the docked conformations of compounds having energy above set cutoff i.e. −25.00 kJ mol−1 predicted by FlexX were identified.19 A total of 225 compounds obtained after applying this filter were meticulously analysed to identify the interactions that contribute significantly in the binding of these compounds with hLigI. All of these compounds docked inside the selected active site and occupy the same position. Among these, 14 compounds were physically available in the CDRI repository at the point of this study and were retrieved for in vitro testing for inhibition of purified ligase activity. The list of these compounds is provided in Table 1.| S. No. | Compounds | Antiligase activity (at 50 μM) (% value) | Docking energy (kJ mol−1) | Structure |
|---|---|---|---|---|
| 1 | S-097/98 | 93.14 | −31.81 | 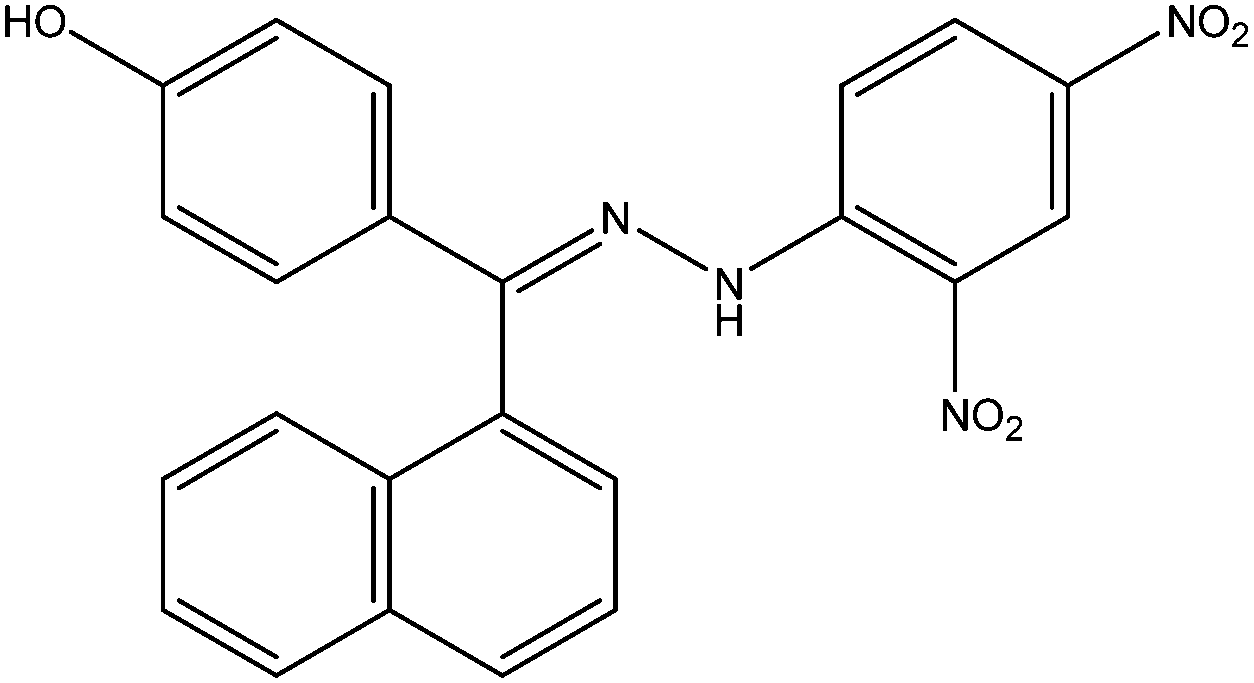 |
| 2 | S-006-12 | 12.2 | −31.00 |  |
| 3 | S-005-1299 | 98.96 | −30.89 | 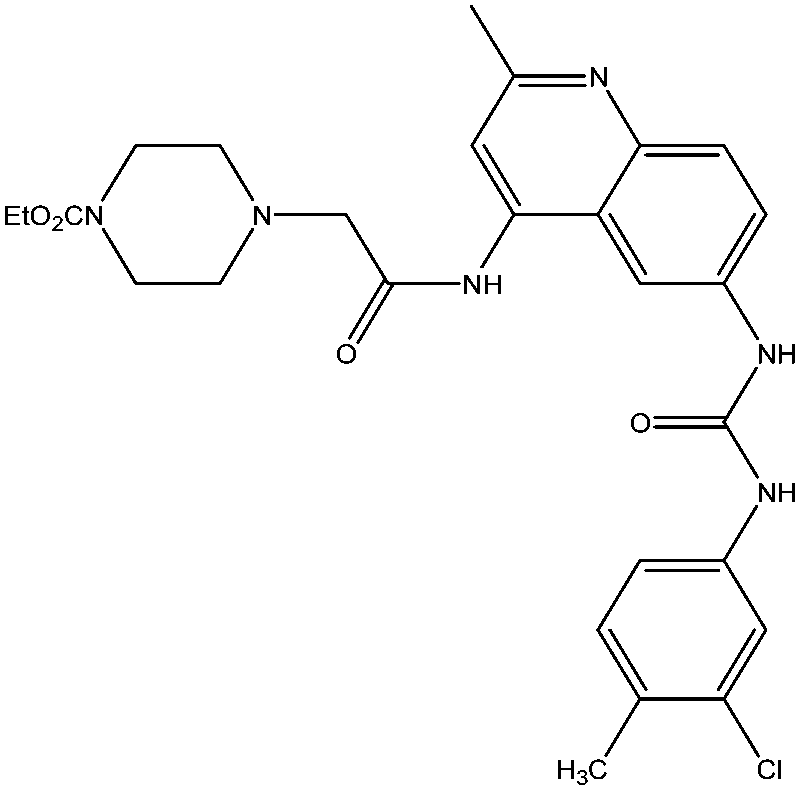 |
| 4 | S-005-1300 | 1.5 | −30.35 |  |
| 5 | S-006-15 | 42.41 | −30.34 |  |
| 6 | S-005-1302 | 10.8 | −29.88 |  |
| 7 | S-005-1298 | 76.18 | −29.39 |  |
| 8 | S-006-1684 | 75.89 | −28.69 |  |
| 9 | S-006-1683 | 98.16 | −28.56 |  |
| 10 | S-006-1690 | 22.35 | −26.80 |  |
| 11 | S-006-1685 | 56.19 | −26.62 |  |
| 12 | S-006-1686 | 64.95 | −26.26 |  |
| 13 | S-006-1161 | 61.09 | −25.93 |  |
| 14 | S-003-1125 | 99.61 | −24.96 |  |
3.2. In vitro screening of predicted hLigI inhibitors
14 compounds obtained from CDRI small molecule library were subjected to in vitro studies. First we performed gel-based, fluorescence-labelled DNA-ligation assays at 50 μM concentration against the purified hLigI enzyme and found that 5 compounds showed >75% inhibition of hLigI activity (Table 1). This was our primary criterion for choosing compounds for further studies. These 5 ligase inhibitors were further tested for antiproliferative activity against various cancer cell lines. We found that only two of the five molecules (S-097/98 and S-005-1299) demonstrated significant antiproliferative activity against the tested cancer cell lines. However, when we tested the two compounds for specific activity against hLigI, we found that only S-097/98 (synthesis scheme and NMR data is provided in ESI material†) was a specific inhibitor of hLigI (Fig. 1) whereas S-005-1299 inhibited all the three human ligases as well as the T4 DNA ligase enzyme (data not shown), hence showing its non-specific nature. This molecule was therefore removed from further studies.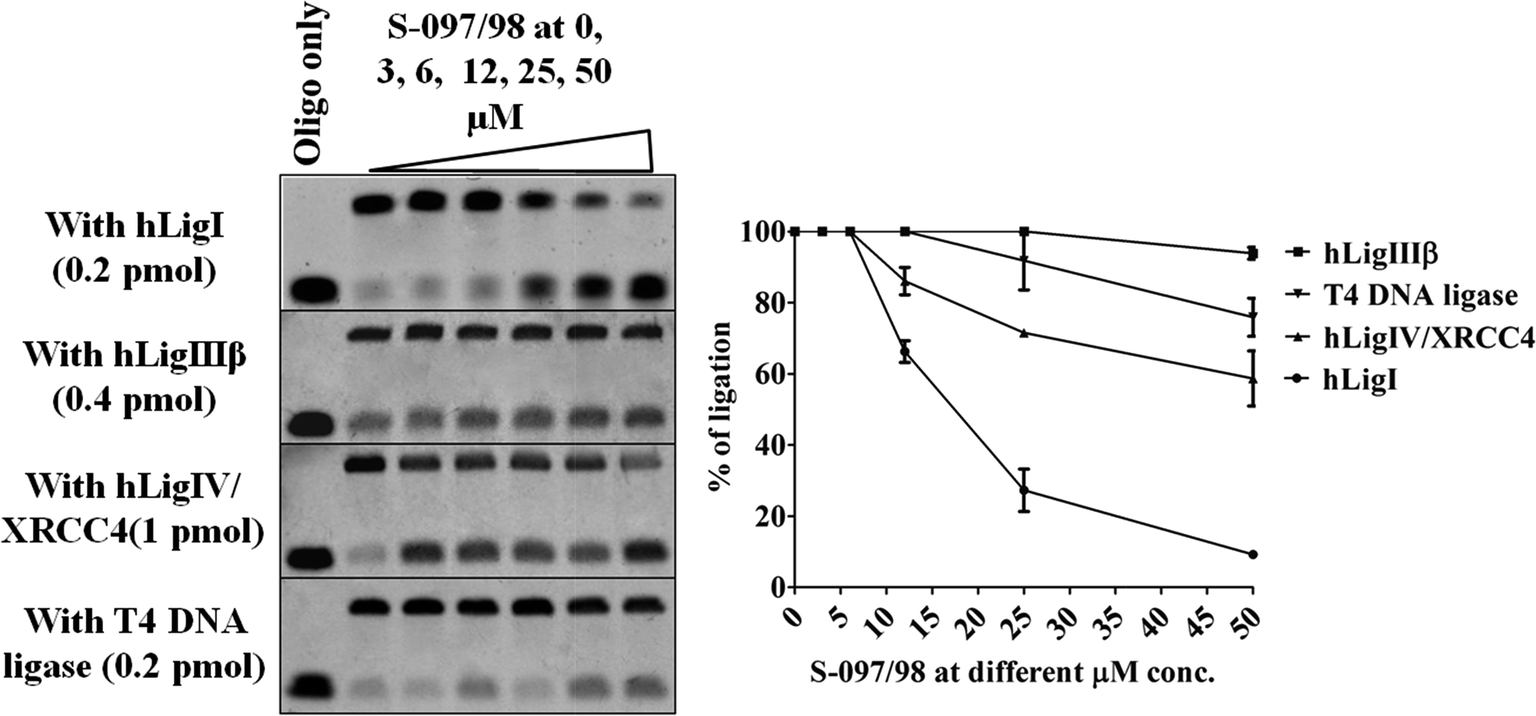 | ||
| Fig. 1 Concentration dependent inhibition of ligation by the compound S-097/98 at 0, 3, 6, 12, 25, 50 μM concentrations against purified ligase proteins (hLigI, hLigIII, hLigIV/XRCC4). The top left panel demonstrates the dose dependent inhibition of hLigI by the compound. The compound has some activity against hLigIV/XRCC4 (left panel, third from top) at higher concentrations, but it is completely inactive against hLigIIIβ (left panel, second from top) and T4 DNA ligases. On the right is the graphical representation of the percentage of ligation observed in the gels at various concentrations of S-097/98. The data shown are mean ± SEM of three independent experiments. | ||
We calculated the IC50 value of S-097/98 in various cancer and normal cell lines as shown in Table 2, and found that it was more active against DLD-1 (colon cancer), HepG2 (liver cancer), and MDA-MB-231 (breast cancer) cell lines with IC50 values <10 μM. Interestingly, the compound S-097/98 was earlier tested for activity against ER positive breast cancer cell lines MCF-7 and ZR-75-1 but the compound was not found to be active against them.47 The reason why the compound S-097/98 exerted different extents of antiproliferative activities on different cancer cell lines may be due to the intrinsic heterogeneity of cancer cells or the interaction of the compound with other targets that enhance the cell death process. Hence the cell killing activity maybe a cumulative effect of the inhibitor on all its targets including hLigI and also depends on the intrinsic differences in the genetic makeup of different cancer cells.48
| Colon cancer (DLD-1) | Liver cancer (HepG2) | Breast cancer (MDA-MB-231) | Human embryonic kidney (HEK-293) | 46Br.1G1 (hLigI deficient human skin fibroblast) |
|---|---|---|---|---|
| 6.78 | 6.93 | 7.02 | 12.6 | >25 |
3.3. Docking of active compound S-097/98 predicts direct binding to hLigI
Fig. 2A shows the pharmacophore mapping of the compound S-097/98. Fig. 2B describes the predicted binding mode of S-097/98 which was found to be the best inhibitor of hLigI. As is evident from Fig. 2B, the compound S-097/98 forms several hydrogen bonds inside the active site of hLigI. Some of the amino acid residues of hLigI that were reported to be involved in hydrogen bonding with DNA in the crystal structure (PDB ID: 1X9N) were also found to interact with S-097/98 by forming hydrogen bonds. Therefore, the binding of S-097/98 would preclude DNA binding as observed in our EMSA results (Fig. 3A). | ||
| Fig. 2 (A) S-097/98 mapped on selected and best validated Hypo1. In Hypo1, the hydrophobic feature is shown in magenta and acceptor atom features are shown in green color. (B) The predicted docking mode of compound S-097/98 within the selected DNA binding site of hLigI crystal structure. The compound is shown in grey sticks and protein in cyan sticks. The black dashed lines represents hydrogen bonds. | ||
 | ||
| Fig. 3 (A) Electrophoretic mobility shift assay (EMSA) performed with S-097/98 at 50, 100, 250 and 500 μM concentrations. The compound S-097/98 reduced the hLigI-nicked DNA complex (lanes 3–6) in a competitive manner. The binding affinity between hLigI and oligo in the presence and absence of inhibitor has been represented quantitatively in the graph. (B) hLigI protein (purified protein in left panel and cell lysate in right panel) was digested with trypsin in the presence and absence of S-097/98. Increasing concentrations of the inhibitor increased the protection of hLigI from trypsin cleavage. The graphs show the relative levels of uncleaved hLigI protein. The data shown are mean ± SEM of three independent experiments. | ||
As shown in Fig. 2, the compound docked well inside the selected active site, which involves major residues contributing to the binding of DNA within the crystal structure. As is evident from the figure, the carbonyl oxygen attached with one of the nitro group of S-097/98 forms a hydrogen bond with Gly 453. The carboxyl oxygen of hydroxyphenyl moiety is engaged in forming hydrogen bonds with Lys770 and Arg451. These residues were observed to interact with DNA by hydrogen bond formation in the crystal structure as well. Additionally, it forms a hydrogen bond with Gly448. The docked pose of S-097/98 also speculates formation of another H-bond via Leu454 (Fig. 2B).
3.4. Specificity of S-097/98 to the activity of different ligases
The specificity of S-097/98 to different human and non-human DNA ligases was tested by performing ligation inhibition assay against all three purified human DNA ligase proteins viz., hLigI, hLigIIIβ, and hLigIV/XRCC4 and the non-human T4 DNA ligase. We found that S-097/98 was specifically active against the hLigI enzyme although at high concentrations the compound has some overlapping activity against the hLigIV/XRCC4 enzyme as well. Moreover, the compound demonstrated a linear dose dependent and selective inhibition activity against hLigI (Fig. 1).3.5. Interaction study of S-097/98 with hLigI protein and DNA
Small molecules can block the ligation activity mainly by two methods, either by direct interaction with the ligase protein and blocking it from accessing the DNA or by binding with the DNA substrate itself and hence occluding the binding of ligase from the DNA non-specifically. We found that S-097/98 directly interacts with hLigI protein and disrupts the interaction between DNA and protein specifically.In order to check the direct binding between hLigI and S-097/98 we performed the Electrophoretic Mobility Shift Assay (EMSA). As seen in Fig. 3A, increasing the concentration of S-097/98 (50–500 μM), reduces the binding affinity between hLigI and nicked DNA substrate (lanes 3–6). Such a loss in binding affinity between hLigI and DNA can occur only due to competition between the inhibitor and substrate DNA for binding with the ligase protein. Of note, in the EMSA experiments, we have used the same molar ratios of the protein and the compound (1:25 to 1:50) as was used in the ligation assays. The apparent increase seen in the inhibitor concentration was due to the higher DNA (2 pmol versus 1 pmol) and protein (0.2 versus 10 pmoles) concentrations that had to be used in the EMSA experiments for proper visualization of the DNA–protein complex. Therefore, apparently higher inhibitor concentrations were necessary to break the complex while maintaining the same molar ratios. Further, since both ligase protein and DNA are present in the reaction here, it is possible that the inhibitor is binding to either of these molecules.
In order to confirm protein binding, we performed additional experiments. In one such experiment, we performed trypsin cleavage of purified hLigI protein as well as hLigI present in the cell lysate (of DLD-1 cells) in the presence and absence of the inhibitor S-097/98 (Fig. 3B). We found that increasing the concentration of inhibitor (to 250 and 500 μM) increases the binding with protein and blocks trypsin cleavage sites present in the hLigI protein. This results in less cleavage of the protein in the presence of inhibitor as compared to control (without inhibitor). We detected the hLigI protein by silver staining in case of purified protein and by Western blotting in case of cell lysate and quantified the band intensities by Image Quant LAS4010 (Fig. 3B). In cell lysate we also checked the trypsin cleavage protection of other replication related proteins like Polδ, RPA, FEN1 and PCNA and we found that hLigI protein got maximum protection in the presence of compound (ESI Fig. 2†), which confirms that compound S-097/98 specifically interact with hLigI protein in the cell lysate.
3.6. Compound S-097/98 does not interact with DNA
In order to rule out DNA binding, we performed DNA intercalation assay and DNaseI cleavage assay in the presence of this (S-097/98) inhibitor. In Fig. 4A, unlike the known DNA intercalator doxorubicin, there was no hindrance seen in the movement of DNA in a 1% agarose gel in the presence of the inhibitor S-097/98 up to 500 μM concentration. This suggests that the compound does not interact with DNA. | ||
| Fig. 4 (A) DNA intercalation assays were performed to demonstrate that there was no hindrance in the movement of DNA (lanes 1–7) in the presence of various concentrations of ampicillin (negative control, lanes 1–3), vehicle control (DMSO, lane 4) and compound (S-097/98, lanes 5–7). However, the addition of the known DNA intercalator doxorubicin (positive control) to the same DNA, led to a hindrance in migration as seen by the higher migrating band in lanes 8–10. (B) Lane 1 shows DNA run in the gel without any DNaseI treatment. 100 ng DNaseI was added to DNA alone (lane 2) or to DNA incubated with either the ligase inhibitor S-097/98 (lanes 3 and 4), doxorubicin (a known DNA intercalator) (lane 5) or ampicillin (negative control) (lane 6). DNaseI cleavage protection was not observed in the presence of the inhibitor S-097/98 but was observed with doxorubicin. | ||
We also performed DNaseI cleavage protection assay (Fig. 4B) to rule out any possibility of interaction between DNA and compound. For this we first incubated the linearized pUC18 DNA with increasing concentrations of S-097/98 (250 and 500 μM), doxorubicin (100 μM) and ampicillin (500 μM) for 30 min, and then cleaved it with DNaseI. We found that unlike doxorubicin the DNA which was incubated with S-097/98 and ampicillin was completely cleaved with DNaseI, which clearly indicates that unlike doxorubicin the compound S-097/98 does not interact with DNA and hence offers no protection from DNaseI digestion.
3.7. Compound S-097/98 induces apoptosis in DLD-1 cell line
Quantitative analysis of the induction of apoptosis was performed in DLD-1 cells using flow-cytometry. DLD-1 cells were treated with different concentrations (7, 10 and 14 μM, where 7 μM is the IC50 concentration for this cell line) of S-097/98 for different time durations (24 and 48 h). Percentage of cells undergoing apoptosis increased with increasing concentrations of S-097/98 and duration of treatment (Fig. 5). After 24 h of treatment, the compound induced 6.9 ± 0.78%, 11.1 ± 1.15% and 32.4 ± 10.4% apoptosis at 7, 10 and 14 μM concentrations respectively whereas, at the same concentrations, 39.16 ± 5.13%, 59.98 ± 2.42% and 63.64 ± 0.46% of DLD-1 cells undergo apoptosis after 48 h of treatment. We have also calculated the percentage of cells undergoing early and late apoptosis when treated with different concentrations of the compound for different time durations. This is represented in the graph in Fig. 5. The significant induction of apoptosis after treatment with S-097/98 is responsible for the cytotoxicity observed in DLD-1 colon cancer cell line.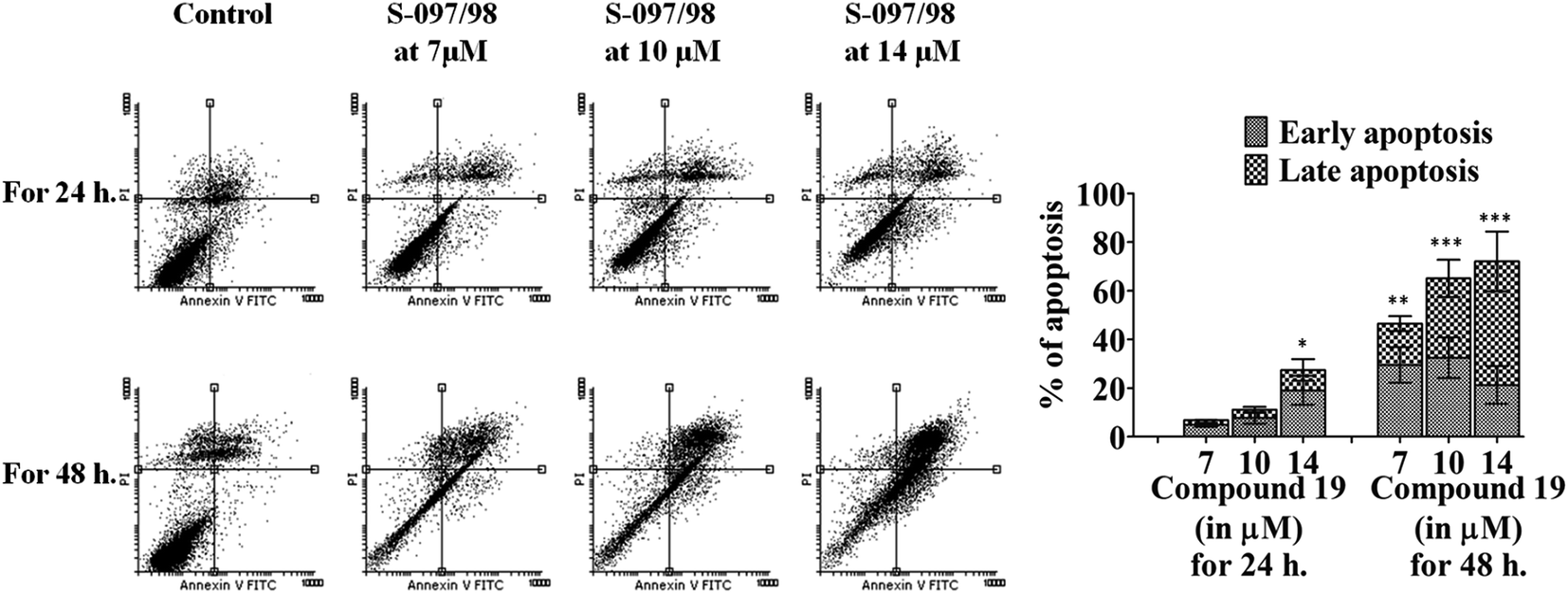 | ||
| Fig. 5 S-097/98 induces apoptosis in DLD-1 cells in a dose dependent manner. DLD-1 cells were treated with varying concentrations (0, 7, 10, 14 μM) of S-097/98 for 24 and 48 h. Cells were then harvested and apoptosis was analysed by FACS analysis. The percentage of cells undergoing early and late apoptosis at different concentrations of the inhibitor are represented graphically. The data shown are mean ± SEM of three independent experiments. Significant difference from control was observed by ANOVA: *P < 0.05, **P < 0.01, ***P < 0.001. | ||
3.8. Compound S-097/98 arrests cell cycle at G2/M phase and increases nuclear size
To study the effect of S-097/98 on cell cycle progression, DLD-1 cells were synchronized by serum starvation for 36 h. After 36 h of synchronization, cells were released into serum containing media and treated with 7 μM (IC50 concentration) of S-097/98 for 0, 12 and 24 h. From Fig. 6A, it is clear that at 0 h, both treated and untreated cells were arrested at G0/G1 phase (59.83 ± 3.5%), followed by 32.46 ± 2.6% cells in the S-phase, and only 7.69 ± 1.02% cells in the G2/M phase. After 12 h of treatment, in control samples, 68.76 ± 2.76% cells were counted in G0/G1 phase and 8.82 ± 0.13% cells were counted in G2/M phase, but in the treated sample 49.55 ± 4.46% and 25.55 ± 5.84% cells were present in G0/G1 and G2/M phase of cell cycle respectively. We found the same pattern of distribution of DLD-1 cells in different phase of cell cycle even after 24 h of treatment. In untreated samples 53.84 ± 11.2% cells were in G0/G1 phase and 14.64 ± 4.84% of cells were in G2/M phase, whereas in treated samples 36.83 ± 6.47% cells were in G0/G1 phase and 36.53 ± 0.81% cells were in G2/M phase. The population in S phase of cell cycle remained almost constant at each time point of treatment. The graph in Fig. 6B, clearly shows that at different time points, S-097/98 increased the population of cells in G2/M phase and decreases the cell population in G0/G1 phase, indicating that this compound blocks the cell cycle at G2/M phase. | ||
| Fig. 6 Cell cycle progression of DLD-1 cells treated with vehicle (DMSO) and compound (S-097/98). (A) DNA histograms clearly show the arrest of cell cycle at the G2/M phase after treatment with S-097/98 for 12 and 24 h at IC50 (7 μM) concentration. The quantitative distribution of cells in different phases of cell cycle is shown graphically. (B) Each bar of the graph is divided into three blocks, upper block represents ‘G2/M phase’, middle block represents ‘S phase’ and lower block represents ‘G0/G1 phase’ of the cell cycle. (C) Shows increased size of nuclei in cells treated with S-097/98 as compared to untreated cells. (D) Shows the differential distribution of nuclear size in treated and control cells at different diameter range (in pixels). (E) Shows the mean area (in pixels) of the nuclei of treated and control cells. (F) The left portion shows FL2-W histogram plots of cytometry analysis of DLD-1 cells treated with S-097/98 and the right side graph shows an increase in nuclear size of treated cells. The data shown are mean ± SEM of three independent experiments performed after 24 h of treatment. Significant difference from control was observed by ANOVA and Student's t test: *P < 0.05, **P < 0.01, ***P < 0.001. | ||
We also checked the effect of S-097/98 on the size of the nucleus of DLD-1 cells. First we grew the DLD-1 cells on coverslips and treated then with S-097/98 at 7 μM (IC50 concentration) for 24 h, stained the cells with propidium iodide (PI) and captured images with a fluorescent microscope. Fig. 6C clearly shows an increase in nuclear size in cells treated with the compound as compared to untreated cells. We analysed the diameter and area of nucleus (in pixels) by the Cell Profiler 2.1.1 image analysis software. From the graph in Fig. 6D, it is clear that in treated sample more cells are found in higher pixels range as compared to control cells. In control sample 306 ± 17 cells, 77 ± 2 cells, 20 ± 2 cells were found in the range of 31–40 pixels, 41–50 pixels and 51–60 pixels of nucleus diameter respectively. Whereas in the treated cells, 270 ± 15 cells, 95 ± 7 cells, and 30 ± 2 cells were found in the range of 31–40 pixels, 41–50 pixels and 51–60 pixels of nuclear diameter respectively. We also calculated the mean area of nucleus (graph from Fig. 6E) of control and treated samples and found that treated cells have larger mean area of nuclei (1170.25 ± 3.75 pixels) as compared to control cells (1077.5 ± 2.5 pixels).
The flow cytometric pulse width analysis also found an enlargement of nuclear size in DLD-1 cells treated with IC50 concentration (7 μM) of S-097/98. The flow cytometer histograms and their corresponding graph (Fig. 6F) clearly show that in the treated sample larger number of cells 47.3 ± 3.7% were distributed in higher FL2-W (refers to nuclear size) range as compared to control cells where 14.8 ± 2.2 cells were distributed in higher FL2-W range. All these observations clearly indicate that the compound S-097/98 increases the nuclear size of DLD-1 cells indicating the arrest of cell cycle.
3.9. Ex vivo demonstration of ligase I inhibition by compound S-097/98
In order to verify whether the compound S-097/98 can target the ligase I activity inside the cells, we checked the ligation activity of DLD-1 cell extract. For this we cultured DLD-1 cells in the presence of the inhibitor at 7 and 14 μM concentrations (or the vehicle control DMSO) for 48 h. We then lysed the cells by brief sonication (2 s pulses × 3 times) and obtained the cell extract and checked the ligation activity of the inhibitor treated and untreated cell extracts (15 μg each). We found that the inhibitor treated samples retained very less ligation activity (<10%) as compared to vehicle (DMSO) treated samples (Fig. 7). | ||
| Fig. 7 A gel picture and graph for the ligation activity of DLD-1 cells treated with compound S-097/98 at different concentrations (0, 7 and 14 μM). Graph is the mean ± SEM of three independent experiments. Significant difference from control was observed by ANOVA: *P < 0.05, **P < 0.01, ***P < 0.001. | ||
3.10. Compound S-097/98 enhances the caspase 3 mediated PARP cleavage due to extensive DNA damage and promotes apoptotic cell death
Inhibition of ligase I activity decreases the nick sealing between DNA ends during replication and repair. The unrepaired DNA gets accumulated in the cells and causes enhanced expression of γH2AX49 (Fig. 8A). During the DNA strand breaks, PARP is activated and helps repair the DNA. However, in case of extensive DNA damage, PARP is over-activated and consumes large amounts of NAD+. In such a situation, the cells in efforts to resynthesize NAD+, may cause massive ATP depletion which may cause necrosis of cells.50 To avoid the necrosis of cells, PARP is inactivated by its cleavage into 24 kDa and 89 kDa fragments. This inactivation of PARP is mediated by activated caspase 3. In Fig. 8B–D, it is clearly shown that increasing the concentration of S-097/98 (from 0 to 7 and 14 μM) increases PARP cleavage and activates caspase 3. These results (enhanced PARP cleavage and caspase 3 activation) directly indicate that the compound S-097/98 enhances caspase 3 mediated PARP cleavage which has been considered a hallmark of apoptosis.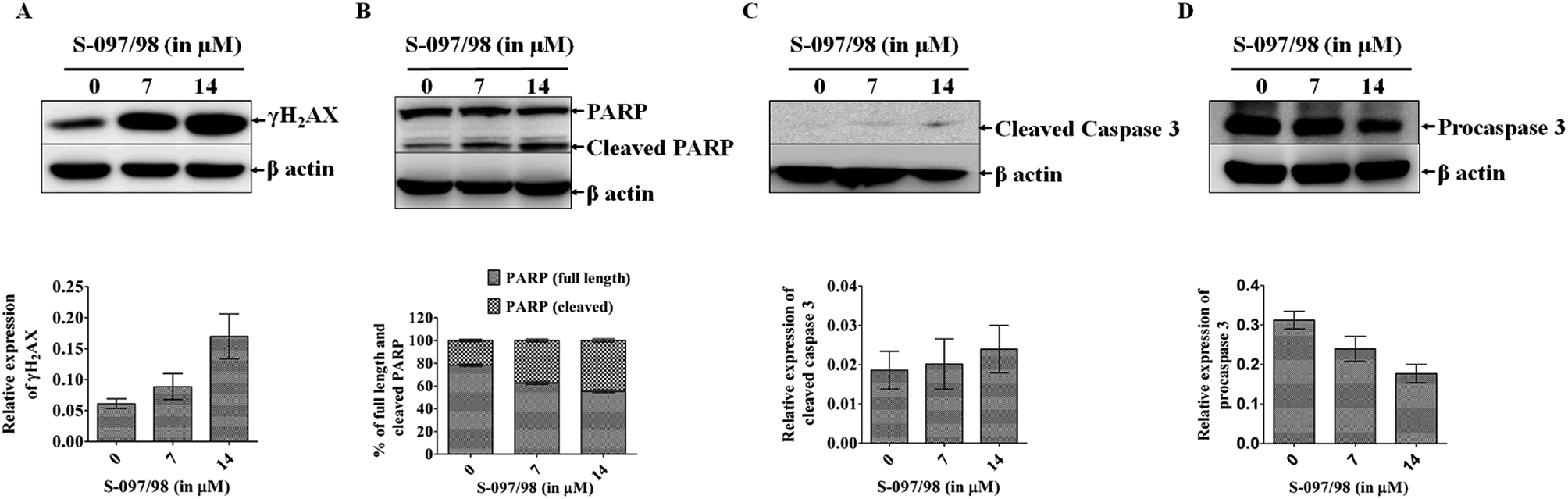 | ||
| Fig. 8 Differential expression level of DNA damage related proteins γH2AX, PARP, and cleaved caspase 3 after treatment with 0, 7 and 14 μM concentrations of S-097/98 for 48 h in DLD-1 cell line. (A) Shows increased expression of γH2AX. The relative expression of γH2AX at different doses of the inhibitor is shown in the graph below the gel. (B) Shows increase in cleavage of PARP, which is a hallmark of apoptosis. The percentage increase in PARP cleavage is shown in the graph below the gel. (C and D) Show increased level of cleaved caspase 3 and decreased level of procaspase 3, both indicators of PARP cleavage and are represented graphically below the gels. The data shown are mean ± SEM of three independent experiments. | ||
4. Conclusion
The major problems associated with cancer therapy are side effects to normal cells and the emergence of drug resistance with time, leading to chemotherapy failure and re-emergence of cancer. To overcome these problems, it is necessary to identify novel targets and their inhibitors for the discovery of new cancer drugs. We predict that human DNA ligases can be targeted for the discovery of new chemotherapeutic agents.17,20 In the present study, we discovered a novel hLigI inhibitor S-097/98 and found that it has antiproliferative activity against ligase expressing cell lines (DLD-1, HepG2, MDA-MB-231 and HEK-293) whereas inactive in ligase I deficient cell line (46Br.1G1) (Table 2). We found that the compound S-097/98 interacts specifically with hLigI (purified as well as cell lysate protein) (Fig. 3 and ESI Fig. 2†) and inhibit their ligation activity (purified as well as cell lysate protein) (Fig. 1 and 7). The compound induces apoptosis in DLD-1 cells and arrests the cell cycle progression in G2/M phase that results in the increase in the nuclear size of cells (Fig. 6). The compound S-097/98 induces the double strand breaks inside the cells and promotes the apoptotic cell death probably by caspase 3 mediated PARP cleavage pathway (Fig. 8). The effects of ligase inhibitors in combination therapy with existing chemotherapeutic and other DNA damaging agents are the subject of further study in our laboratory.Acknowledgements
The authors acknowledge CSIR-CDRI, Govt. of India for financial and infrastructural support. Financial support is also acknowledged from Department of Biotechnology (DBT), Govt. of India (Grant-BT/PR6421/GBD/27/436/2012), the Department of Science and Technology (DST), Govt. of India (Grant-SB/FT/LS-163/2012) and CSIR network project GENESIS (BSC0121). The authors are also thankful to Mr A. L. Vishwakarma for his support during flowcytometry experiments. DKS, ALD, SK and MS are thankful for their Senior Research fellowships from Council of Scientific and Industrial Research (CSIR), and Indian Council of Medical Research, and UGC New Delhi, India. The manuscript bears the CDRI manuscript number 9339.References
- S. P. Jackson and J. Bartek, Nature, 2009, 461, 1071–1078 CrossRef CAS PubMed.
- B. B. Zhou and S. J. Elledge, Nature, 2000, 408, 433–439 CrossRef CAS PubMed.
- A. Sancar, L. A. Lindsey-Boltz, K. Unsal-Kacmaz and S. Linn, Annu. Rev. Biochem., 2004, 73, 39–85 CrossRef CAS PubMed.
- G. M. Kupfer, Yale J. Biol. Med., 2013, 86, 491–497 CAS.
- J. M. Daley, Y. Kwon, H. Niu and P. Sung, Yale J. Biol. Med., 2013, 86, 453–461 CAS.
- J. M. Murray and A. M. Carr, Nat. Rev. Mol. Cell Biol., 2008, 9, 177–182 CrossRef CAS PubMed.
- E. Pastwa and J. Blasiak, Acta Biochim. Pol., 2003, 50, 891–908 CAS.
- Z. Wang, X. Wu and E. C. Friedberg, J. Biol. Chem., 1997, 272, 24064–24071 CrossRef CAS PubMed.
- N. G. Jaspers and J. H. Hoeijmakers, Curr. Biol., 1995, 5, 700–702 CrossRef CAS PubMed.
- A. P. Eker, C. Quayle, I. Chaves and G. T. van der Horst, Cell. Mol. Life Sci., 2009, 66, 968–980 CrossRef CAS PubMed.
- A. A. Larrea, S. A. Lujan and T. A. Kunkel, Cell, 2010, 141, 730 CrossRef PubMed.
- R. E. Verdun and J. Karlseder, Nature, 2007, 447, 924–931 CrossRef CAS PubMed.
- E. Weterings and D. J. Chen, J. Cell Biol., 2007, 179, 183–186 CrossRef CAS PubMed.
- L. H. Pearl, A. C. Schierz, S. E. Ward, B. Al-Lazikani and F. M. Pearl, Nat. Rev. Cancer, 2015, 15, 166–180 CrossRef CAS PubMed.
- S. Zhong, X. Chen, X. Zhu, B. Dziegielewska, K. E. Bachman, T. Ellenberger, J. D. Ballin, G. M. Wilson, A. E. Tomkinson and A. D. MacKerell Jr, J. Med. Chem., 2008, 51, 4553–4562 CrossRef CAS PubMed.
- X. Chen, S. Zhong, X. Zhu, B. Dziegielewska, T. Ellenberger, G. M. Wilson, A. D. MacKerell Jr and A. E. Tomkinson, Cancer Res., 2008, 68, 3169–3177 CrossRef CAS PubMed.
- A. E. Tomkinson, T. R. Howes and N. E. Wiest, Transl. Cancer Res., 2013, 2(3), pii: 1219 Search PubMed.
- M. Srivastava, M. Nambiar, S. Sharma, S. S. Karki, G. Goldsmith, M. Hegde, S. Kumar, M. Pandey, R. K. Singh, P. Ray, R. Natarajan, M. Kelkar, A. De, B. Choudhary and S. C. Raghavan, Cell, 2014, 151, 1474–1487 CrossRef PubMed.
- S. Krishna, D. K. Singh, S. Meena, D. Datta, M. I. Siddiqi and D. Banerjee, J. Chem. Inf. Model., 2014, 54, 781–792 CrossRef CAS PubMed.
- D. K. Singh, S. Krishna, S. Chandra, M. Shameem, A. L. Deshmukh and D. Banerjee, Med. Res. Rev., 2014, 34, 567–595 CrossRef CAS PubMed.
- M. Shameem, R. Kumar, S. Krishna, C. Kumar, M. I. Siddiqi, B. Kundu and D. Banerjee, Chem.-Biol. Interact., 2015, 237, 115–124 CrossRef CAS PubMed.
- D. Mandalapu, D. K. Singh, S. Gupta, V. M. Balaramnavar, M. Shafiq, D. Banerjee and V. L. Sharma, RSC Adv., 2016, 6, 26003–26018 RSC.
- M. Pandey, S. Kumar, G. Goldsmith, M. Srivastava, S. Elango, M. Shameem, D. Bannerjee, B. Choudhary, S. S. Karki and S. C. Raghavan, Mol. Carcinog., 2016 DOI:10.1002/mc.22516.
- S. Soderhall, Eur. J. Biochem., 1975, 51, 129–136 CrossRef CAS PubMed.
- I. R. Lehman, Science, 1974, 186, 790–797 CAS.
- S. Soderhall and T. Lindahl, J. Biol. Chem., 1973, 248, 672–675 CAS.
- Y. F. Wei, P. Robins, K. Carter, K. Caldecott, D. J. Pappin, G. L. Yu, R. P. Wang, B. K. Shell, R. A. Nash and P. Schar, et al., Mol. Cell. Biol., 1995, 15, 3206–3216 CrossRef CAS PubMed.
- D. E. Barnes, L. H. Johnston, K. Kodama, A. E. Tomkinson, D. D. Lasko and T. Lindahl, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 6679–6683 CrossRef CAS.
- P. Robins and T. Lindahl, J. Biol. Chem., 1996, 271, 24257–24261 CrossRef CAS PubMed.
- T. Ellenberger and A. E. Tomkinson, Annu. Rev. Biochem., 2008, 77, 313–338 CrossRef CAS PubMed.
- D. S. Levin, W. Bai, N. Yao, M. O'Donnell and A. E. Tomkinson, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12863–12868 CrossRef CAS.
- R. Prasad, R. K. Singhal, D. K. Srivastava, J. T. Molina, A. E. Tomkinson and S. H. Wilson, J. Biol. Chem., 1996, 271, 16000–16007 CrossRef CAS PubMed.
- A. B. Robertson, A. Klungland, T. Rognes and I. Leiros, Cell. Mol. Life Sci., 2009, 66, 981–993 CrossRef CAS PubMed.
- G. L. Dianov, Am. J. Cancer Res., 2011, 1, 845–851 CAS.
- L. Liang, L. Deng, S. C. Nguyen, X. Zhao, C. D. Maulion, C. Shao and J. A. Tischfield, Nucleic Acids Res., 2008, 36, 3297–3310 CrossRef CAS PubMed.
- J. Moser, H. Kool, I. Giakzidis, K. Caldecott, L. H. Mullenders and M. I. Fousteri, Mol. Cell, 2007, 27, 311–323 CrossRef CAS PubMed.
- T. E. Wilson, U. Grawunder and M. R. Lieber, Nature, 1997, 388, 495–498 CrossRef CAS PubMed.
- A. Montecucco, G. Biamonti, E. Savini, F. Focher, S. Spadari and G. Ciarrocchi, Nucleic Acids Res., 1992, 20, 6209–6214 CrossRef CAS PubMed.
- D. Sun, R. Urrabaz, M. Nguyen, J. Marty, S. Stringer, E. Cruz, L. Medina-Gundrum and S. Weitman, Clin. Cancer Res., 2001, 7, 4143–4148 CAS.
- GASP, Sybyl, 7.1 edn, Tripos, Inc., St. Louis, MO, 2004 Search PubMed.
- UNITY, Sybyl, 7.1 edn, Tripos, Inc., St. Louis, MO, 2004 Search PubMed.
- J. M. Pascal, P. J. O'Brien, A. E. Tomkinson and T. Ellenberger, Nature, 2004, 432, 473–478 CrossRef CAS PubMed.
- M. Rarey, B. Kramer, T. Lengauer and G. Klebe, J. Mol. Biol., 1996, 261, 470–489 CrossRef CAS PubMed.
- B. Lomenick, R. Hao, N. Jonai, R. M. Chin, M. Aghajan, S. Warburton, J. Wang, R. P. Wu, F. Gomez, J. A. Loo, J. A. Wohlschlegel, T. M. Vondriska, J. Pelletier, H. R. Herschman, J. Clardy, C. F. Clarke and J. Huang, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21984–21989 CrossRef CAS PubMed.
- K. Kang, S. B. Lee, J. H. Yoo and C. W. Nho, Biotechnol. Lett., 2010, 32, 1045–1052 CrossRef CAS PubMed.
- K. Kang, S. H. Oh, J. H. Yun, E. H. Jho, J. H. Kang, D. Batsuren, J. Tunsag, K. H. Park, M. Kim and C. W. Nho, Neoplasia, 2010, 13, 1043–1057 CrossRef.
- J. Pandey, R. Pal, A. Dwivedi and K. Hajela, Arzneimittelforschung, 2002, 52, 39–44 CAS.
- S. Jaeger, M. Duran-Frigola and P. Aloy, Mol. Cancer, 2015, 14, 40 CrossRef PubMed.
- L. J. Kuo and L. X. Yang, In Vivo, 2008, 22, 305–309 CAS.
- M. Los, M. Mozoluk, D. Ferrari, A. Stepczynska, C. Stroh, A. Renz, Z. Herceg, Z. Q. Wang and K. Schulze-Osthoff, Mol. Biol. Cell, 2002, 13, 978–988 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra22364h |
| ‡ Present address: Department of Chemistry, Govt. Raza Post Graduate College, Rampur-244901, India. |
| This journal is © The Royal Society of Chemistry 2016 |