
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural and electronic determinants of flavonoid binding to human serum albumin: an extensive ligand-based study
Hrvoje
Rimac
a,
Željko
Debeljak
bc,
Davor
Šakić
a,
Tin
Weitner
a,
Mario
Gabričević
a,
Valerije
Vrček
a,
Branka
Zorc
a and
Mirza
Bojić
*a
aUniversity of Zagreb, Faculty of Pharmacy and Biochemistry, Department of Medicinal Chemistry, A. Kovačića 1, HR-10000 Zagreb, Croatia. E-mail: mbojic@pharma.hr
bClinical Hospital Center Osijek, Osijek, Croatia
cJ.J. Strossmayer University of Osijek, Faculty of Medicine, Osijek, Croatia
First published on 3rd August 2016
Abstract
Flavonoids are ubiquitous plant metabolites that interfere with different biological processes in the human organism. After absorption they bind to human serum albumin (HSA), the most abundant carrier protein in the blood which also binds various hormones and drugs. Binding of flavonoids to HSA may impact their distribution, influencing the active concentration in the blood. To determine the most prominent features responsible for binding of 20 different flavonoid aglycones to the IIA region of HSA, in vitro fluorescence measurements and density functional theory (DFT) calculations were conducted. These results were then integrated to elucidate structure–affinity relationships. The presented results reveal that flavones and flavonoles bind most strongly to the IIA region of HSA. There are several electronic and structural determinants associated with flavonoid binding to this HSA region: high C3 nucleophilicity and partial charge of O4, high HOMO and LUMO energies, and coplanarity of AC and B rings. Both steric and electronic characteristics of flavonoids have a great impact on their binding to HSA, with hydrogen donor and acceptor properties and coplanarity being the most prominent.
1. Introduction
Human serum albumin (HSA) is the most abundant protein in human plasma (60%, w/w),1 which structure has been determined crystallographically. It is a 585-residue monomeric protein comprised of three homologous domains (I–III), each of which is composed of two subdomains (A and B) (Fig. 1).2 These domains play a central role in binding of various endo- and exogenous compounds, particularly hydrophobic organic anions of medium size (100 to 600 Da), e.g. bilirubin, long-chain fatty acids, hematin, thyroxin.3 Many drugs bind to one of the two primary binding sites, located in subdomains IIA and IIIA (Sudlow sites I and II, respectively), with IIA being the most prominent one.4,5 The IIA subdomain appears to be spacious and is comprised of several individual binding sites which can accommodate ligands characterized by very different chemical structures. The IIIA site is smaller and less flexible and thus can accommodate only structurally similar ligands.6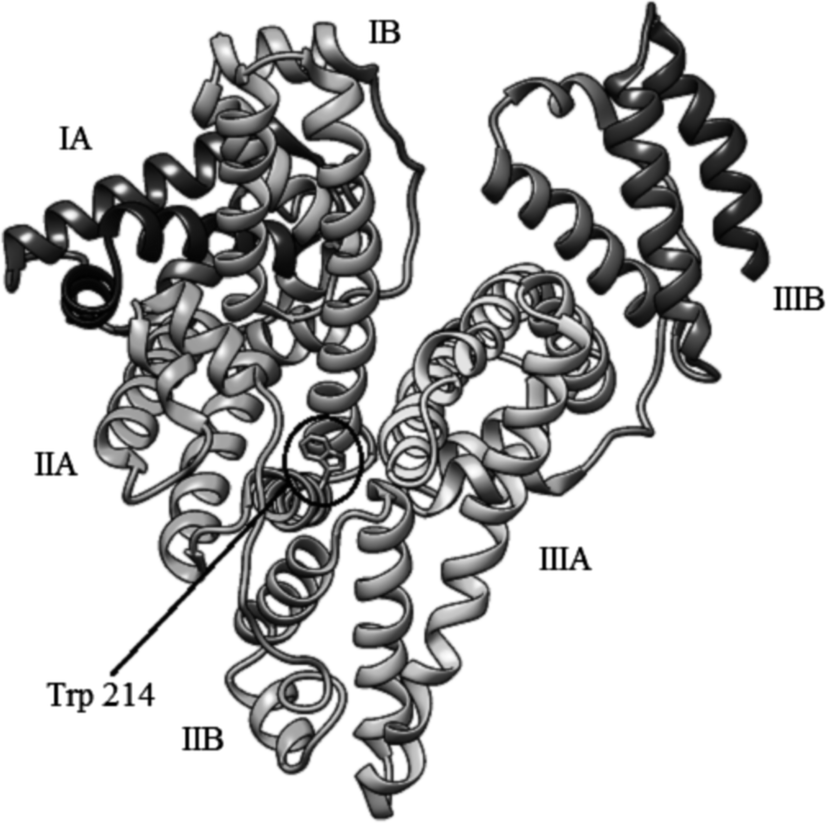 | ||
| Fig. 1 Structure of HSA (PDB entry 1AO6). | ||
A large number of drugs characterized by narrow therapeutic windows, such as warfarin,7 amlodipine,8 various antiepileptic drugs9,10etc. bind to HSA. It has been demonstrated that their binding significantly influences their distribution, free blood concentration and metabolism,9,11 primarily in kidney and liver patients.12 Apart from drugs, various other exogenous compounds bind to HSA as well, such as flavonoids. Flavonoids are a group of phenolic compounds, ubiquitous in fruits and vegetables.13 They have many salutary properties, among which antioxidative properties are best described.14 They also help to prevent lipid peroxidation15 and cardiovascular diseases.16–18 Once they reach circulation, most of these compounds bind in subdomain IIA of HSA,19–22 with flavanones being an exception.23 When flavonoid binds to the same site as a hormone, drug or even a toxin;24 the displaced compound's free concentration in blood may increase. This can lead to changes in pharmacodynamics and pharmacokinetics of the compound in question, such as enhanced pharmacological or adverse effects and its faster elimination.
Binding of flavonoids to HSA has been extensively studied using different fluorescence spectroscopy techniques,19,20,23,25–30 circular dichroism,21,25,27,29,30 and molecular modeling.21,25–30 Binding of ligands in the IIA subdomain of HSA can be monitored by measuring the fluorescence intensity of the Trp 214 residue. Change in tryptophan fluorescence intensity is observed when a ligand binds to HSA in its vicinity. The observed change in fluorescence enables the calculation of HSA–ligand complexes stability constants and also the distance between the ligand and Trp 214.31
The IIA subdomain of HSA is comprised of a neutral hydrophobic cavity and a positively charged hydrophilic part.2,32 At physiological pH, the B ring of flavonoids (Fig. 2) possess a partial negative charge, while chromene part (rings A and C) is hydrophobic, creating ideal conditions for flavonoid binding.33 This has been modeled by docking studies of flavone luteolin,27 flavonol quercetin,21 and isoflavone daidzein.25,26,34 These studies only included few structurally similar flavonoids, so general structure–affinity relationship of HSA–flavonoid binding have not yet been determined.
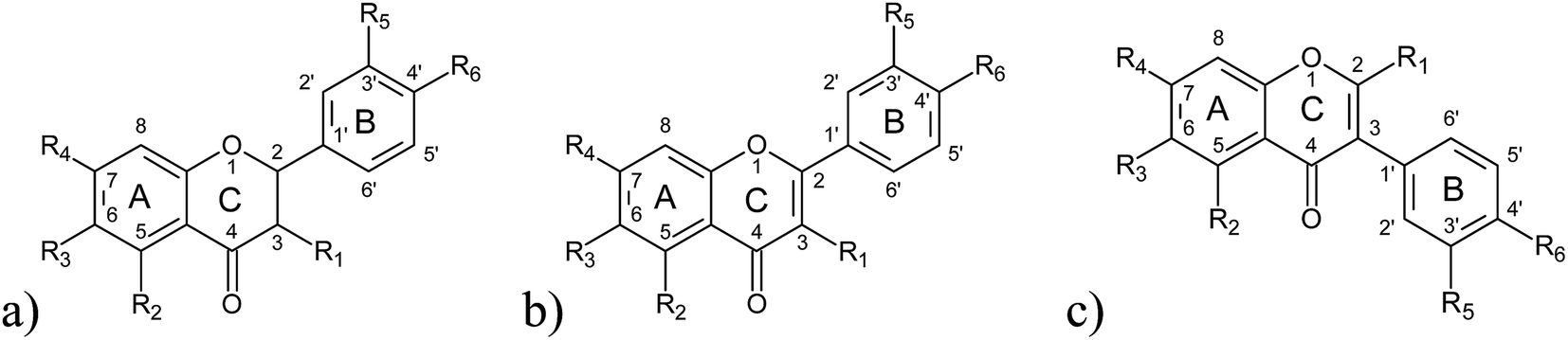 | ||
| Fig. 2 Structural formulae of investigated classes of flavonoids: (a) flavanones, (b) flavones and flavonols, (c) isoflavones. | ||
To determine a common structure–affinity relationship of HSA–flavonoid binding 20 flavonoid aglycones were chosen in this study. In vitro binding constants of these compounds were determined using fluorescence spectrophotometry and their geometries and electronic properties were calculated at the density functional theory (DFT) level of theory. B3LYP model was used to determine electronic and structural features of flavonoids that may influence their binding to the IIA subdomain and effects of solvation have been evaluated. To the best of our knowledge, this is the most extensive study of HSA–flavonoid binding. Significance of obtained results is more accentuated by the fact that in the recent years flavonoid scaffolds have been recognized as privileged scaffolds in drug discovery.35,36
2. Materials and methods
2.1. Materials
Fatty acid free HSA was purchased from Sigma-Aldrich, USA (purity ≥ 96%). Flavonoids were obtained from Extrasynthèse, France (chrysin dimethylether, diosmetin, fisetin, formononetin, genistein, pinocembrin-7-methylether, prunetin, sakuranetin, and tamarixetin), ChromaDex, USA (3,6-dihydroxyflavone, 3,7-dihydroxyflavone, 6-hydroxyflavone, 7-hydroxyflavone, and flavanone), Sigma-Aldrich, USA (apigenin, chrysin, flavone, galangin, and quercetin), and BioChemika, Switzerland (rhamnetin). All flavonoid standards had a specified purity of ≥98%.2.2. In vitro fluorescence measurements
Albumin was dissolved daily before measurements in Dulbecco's phosphate-buffered saline (137 mM sodium chloride, 2.7 mM potassium chloride, 8.1 mM disodium hydrogen phosphate, 1.47 mM potassium dihydrogen phosphate).37 Flavonoids were dissolved in DMSO. A series of solutions were made for each HSA–flavonoid combination: the concentration of HSA was held constant at 1 μM and the flavonoid concentration ranged 0.03–10 μM. In all experiments the maximal DMSO concentration was 4% v/v. The effect of DMSO as a co-solvent was annulated by adding a small aliquot to a pure HSA solution.Steady-state fluorescence spectra were recorded on the OLIS RSM 1000F spectrofluorimeter (Olis Inc., Bogart, GA, USA) equipped with a thermostated cell holder at 25 °C. Hellma Analytics 105.253-QS fluorescence cells with a light path of 10 × 2 mm (excitation × emission) were used. Excitation wavelength was 280 nm (albumin absorption maximum) and the emission spectra were recorded in the range 310–370 nm, where only HSA has fluorescent properties, with the observed maximum at 340 nm. All studies were performed in duplicate at 25 °C using 1.24 mm excitation and emission slit widths. All solutions were analyzed after 2 hours incubation period.
In the studied wavelength range flavonoids absorb light as well. Based on the molar absorbance coefficients at 280 and 340 nm, the inner filter effect cannot be neglected.38 The observed fluorescence, Fobs, has been corrected to Fcorr according to eqn (1), where Aex = ε280 × c × l is absorbance at the excitation wavelength (c is flavonoid concentration and l = 1 cm) and Aem = ε340 × c × l is absorbance at the emission wavelength (l = 0.2 cm):
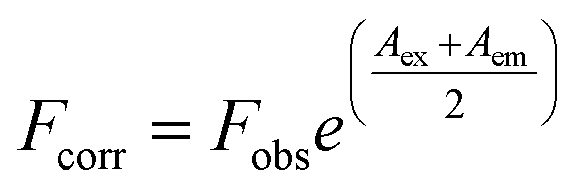 | (1) |
2.3. Calculation of binding constants
Each spectrum obtained during titration is an average of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 fluorescence spectra measured in 10 seconds. Equilibrium constants were calculated by a global fit at all wavelengths with SPECFIT39–42 software. A single significant spectrally active species was suggested by singular value decomposition analysis and attributed to the known spectrum of HSA. This analysis also suggested 1-to-1 complex formation and did not indicate any higher order complexes. Therefore, the proposed binding model is given by eqn (2) and (3), where KA is the association constant of the complex:
000 fluorescence spectra measured in 10 seconds. Equilibrium constants were calculated by a global fit at all wavelengths with SPECFIT39–42 software. A single significant spectrally active species was suggested by singular value decomposition analysis and attributed to the known spectrum of HSA. This analysis also suggested 1-to-1 complex formation and did not indicate any higher order complexes. Therefore, the proposed binding model is given by eqn (2) and (3), where KA is the association constant of the complex: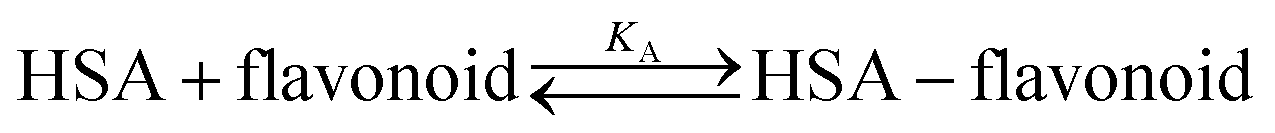 | (2) |
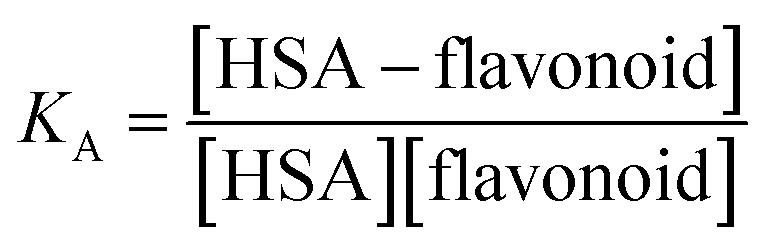 | (3) |
The association constant values calculated from fluorescence titrations for the suggested HSA–flavonoid complexes are given in mol−1 dm3. Higher values of KA indicate stronger binding and higher complex stability. In all cases, logKA was calculated.
2.4. DFT calculations
Quantum chemical calculations on flavonoids were performed using the Gaussian09 program.43 All structures (neutral and anionic forms, where available) were optimized with B3LYP functional,44,45 using Pople's 6-31G(d) and 6-311++G(d,p) basis sets.46,47 B3LYP functional was selected as one of the most used functional for initial screening of large molecules, due to good precision/performance ratio. Flavonoid structures were recently explored using this method.48,49 Analytical vibrational analysis was performed at the corresponding levels to characterize each stationary point on the potential energy surface as a minimum (NImag = 0).Structures of flavones, flavonols, and isoflavones are simple in terms of conformational flexibility. The main parameter which governs the structural diversity is a dihedral angle along C2–C1′ (flavones and flavonols) or C3–C1′ (isoflavones). Dihedral scans were calculated through relaxed scanning of potential energy surfaces at 30° intervals. In this scanning procedure, for each change of the corresponding torsional angle, the structure is fully optimized for all degrees of freedom, affording unconstrained minima to be located. In all global minima, aka lowest lying energy minimum for each flavonoid structure, corresponding dihedral angles are around 0°. An additional contribution to the coplanarity between AC and B rings of flavones, flavonols, and isoflavones comes from hydroxyl/methoxy groups which are rotated to form intramolecular hydrogen bonds, in corresponding global minima. All global minima located at the B3LYP/6-311++G(d,p) level are in agreement with calculated data reported earlier.50,51
In case of flavanones the same conformational procedure was followed, with special attention to the pyranone ring C which adopts a slightly modified sofa conformation in all global minimum structures. These results match the earlier conformational searches on flavanones.52,53
Boltzmann distribution was used to identify the conformers that have more than 5% ratio in total population (based on Gibbs free energy and room temperature), and those structures have been included in further analysis.
To properly model solvent effects in aqueous medium (dielectric constant of ε = 78.4), three different methods were included: (a) implicit solvation using Solvation Model based on Density (SMD),54 where water is treated as a continuum, (b) explicit solvation that includes one water molecule placed in the vicinity of the most electronegative atom, and (c) supramolecular approach where both implicit and explicit solvation is included.55 Due to high similarity between in vacuo and various solvation models, in the remainder of the text only the in vacuo and the explicit solvation are commented. Explicit solvation can also be considered as complexation, and since the IIA binding site is highly hydrophobic,2 it represents the most likely influence of water on the HSA–flavonoid system. If not mentioned otherwise, results refer to in vacuo data.
Natural bond orbital (NBO) analysis was performed using NBO 3.1 program56 included in the Gaussian09 package. NBO analysis (NPA values) and population analysis (Mulliken and APT values) of substituent functional groups were obtained by summarizing individual charge/population on each atom in the functional group. Fukui parameters (electrophilicity and nucleophilicity) were calculated using single-point NBO and population analysis calculations on corresponding N − 1 and N + 1 (N equals number of electrons) systems.
To facilitate quantitative comparison between different sites, the condensed Fukui function57–59 based on atomic charges was calculated. NPA charges from NBO analysis were used. Charges (q) were calculated for all flavonoids in their N, N + 1, and N − 1 electrons states to obtain the condensed f− and f+ descriptors according to equations for the nucleophilicity (4) and electrophilicity (5):
| f−A = qA(N) − qA(N − 1) | (4) |
| f+A = qA(N + 1) − qA(N) | (5) |
All statistical calculations have been done by Statistica 7.0 (Statsoft, USA). p is a measure of statistical significance: p values below 0.05 were considered as significant throughout the text. Univariate linear regression and 2D contour plots have been used in some instances to describe relationship between HSA binding affinity and different structural and electronic features of flavonoids.
3. Results and discussion
3.1. Determination of binding constants
General structural formulas (Fig. 2) and binding constants of the investigated flavonoids are shown below (Table 1). In general, flavanones are characterized by the lowest binding constants, followed by isoflavones. Flavones and flavonols have the highest values of binding constants. Determination of apigenin binding constant (Table 1, entry 6) is shown in Fig. 3. Binding constants for other flavonoids were calculated in the same manner. Most of the binding constants are in accordance with data from previously published studies, with a few exceptions: for some flavonoids literature data could not be found. For 3,6-dihydroxyflavone and tamarixetin, there is a slight discrepancy with literature data, which could be explained by different techniques used to obtain their binding constants. The literature binding constant of 3,6-dihydroxyflavone is lower than the one obtained in this study, which can be explained by fact that 3,6-dihydroxyflavone is also slightly fluorescent under the same conditions, i.e. fluorescence quenching and displacement of quercetin could produce falsely decreased values. In the case of tamarixetin, discrepancies can be explained by the fact that albumins from two different species were used.19 Greater differences are detected for galangin and formononetin. Published galangin binding constant comes from warfarin displacement experiments, where decrease in warfarin anisotropy, i.e. higher rotational freedom of warfarin was taken as a proof of warfarin displacement. As shown by Yamasaki et al.,60 changes in rotational freedom are not necessarily equal to ligand displacement or binding. Similar reasoning can be applied in the case of formononetin. In some cases fluorescence quenching studies are performed at excitation wavelength of 280 nm (where both tryptophan and tyrosine residues absorb) and in another at 295 nm (where only the warfarin residue absorbs), which can also lead to discrepancies. These differences are not so pronounced, but even small discrepancies may compromise relative comparison of ligands, especially if their binding constants are not very different. Therefore, a study of binding constants conducted under the uniform conditions is a necessary prerequisite for a reliable determination of the structure–affinity relationship.| # | Flavonoid | R 1 | R 2 | R 3 | R 4 | R 5 | R 6 | K A [M−1] | K A [M−1] from literature | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| a Calculations are based on competitive binding with warfarin using fluorescence anisotropy approach. b Calculated for bovine serum albumin (BSA) using fluorescence quenching approach. c Calculations are based on competitive binding with quercetin using fluorescence quenching approach. N/A not available. | ||||||||||
| Flavanones | ||||||||||
| 1 | Flavanone | H | H | H | H | H | H | (5.25 ± 0.79) × 103 | N/A | N/A |
| 2 | Pinocembrin-7-methylether | H | OH | H | OCH3 | H | H | (2.19 ± 0.09) × 104 | N/A | N/A |
| 3 | Sakuranetin | H | OH | H | OCH3 | H | OH | (2.19 ± 0.04) × 104 | N/A | N/A |
|
||||||||||
| Flavones | ||||||||||
| 4 | 6-Hydroxyflavone | H | H | OH | H | H | H | (1.58 ± 0.60) × 104 | N/A | N/A |
| 5 | 7-Hydroxyflavone | H | H | H | OH | H | H | (1.95 ± 0.10) × 105 | 9.44 × 104 to 3.82 × 105 | 61 and 62 |
| 6 | Apigenin | H | OH | H | OH | H | OH | (1.32 ± 0.05) × 105 | 9.85 × 104 to 1.95 × 106 | 63 and 64 a |
| 7 | Chrysin | H | OH | H | OH | H | H | (1.95 ± 0.08) × 105 | 1.82 × 105 to 3.09 × 106 | 62, 64 a and 65 |
| 8 | Chrysin dimethylether | H | OCH3 | H | OCH3 | H | H | (2.95 ± 0.15) × 104 | N/A | N/A |
| 9 | Diosmetin | H | OH | H | OH | OH | OCH3 | (8.91 ± 0.18) × 104 | 5.28 × 104 to 1.18 × 105 | 19 b and 66 |
| 10 | Flavone | H | H | H | H | H | H | (6.17 ± 0.56) × 104 | 4.96 × 104 | 62 |
|
||||||||||
| Flavonoles | ||||||||||
| 11 | 3,6-Dihydroxyflavone | OH | H | OH | H | H | H | (7.41 ± 0.15) × 104 | 5.28 × 104 | 67 c |
| 12 | 3,7-Dihydroxyflavone | OH | H | H | OH | H | H | (1.66 ± 0.05) × 105 | (1.51 ± 0.23) × 105 | 68 |
| 13 | Fisetin | OH | H | H | OH | OH | OH | (1.20 ± 0.05) × 105 | (1.38 ± 0.02) × 105 | 69 |
| 14 | Galangin | OH | OH | H | OH | H | H | (2.34 ± 0.09) × 104 | (3.80 ± 0.91) × 106 | 64 a |
| 15 | Quercetin | OH | OH | H | OH | OH | OH | (1.70 ± 0.03) × 105 | 3.1 × 104 to 3.31 × 105 | 19,b34, 64,a65 and 70–72 |
| 16 | Rhamnetin | OH | OH | H | OCH3 | OH | OH | (1.29 ± 0.03) × 105 | N/A | N/A |
| 17 | Tamarixetin | OH | OH | H | OH | OH | OCH3 | (2.34 ± 0.07) × 104 | (7.46 ± 0.30) × 104 | 19 b |
|
||||||||||
| Isoflavones | ||||||||||
| 18 | Formononetin | H | H | H | OH | H | OCH3 | (2.14 ± 0.11) × 104 | 1.60 × 105 | 73 |
| 19 | Genistein | H | OH | H | OH | H | OH | (4.90 ± 0.10) × 104 | 1.14 × 104 to 1.5 × 105 | 19,b25, 71 and 74 |
| 20 | Prunetin | H | OH | H | OCH3 | H | OH | (3.80 ± 0.14) × 104 | N/A | N/A |
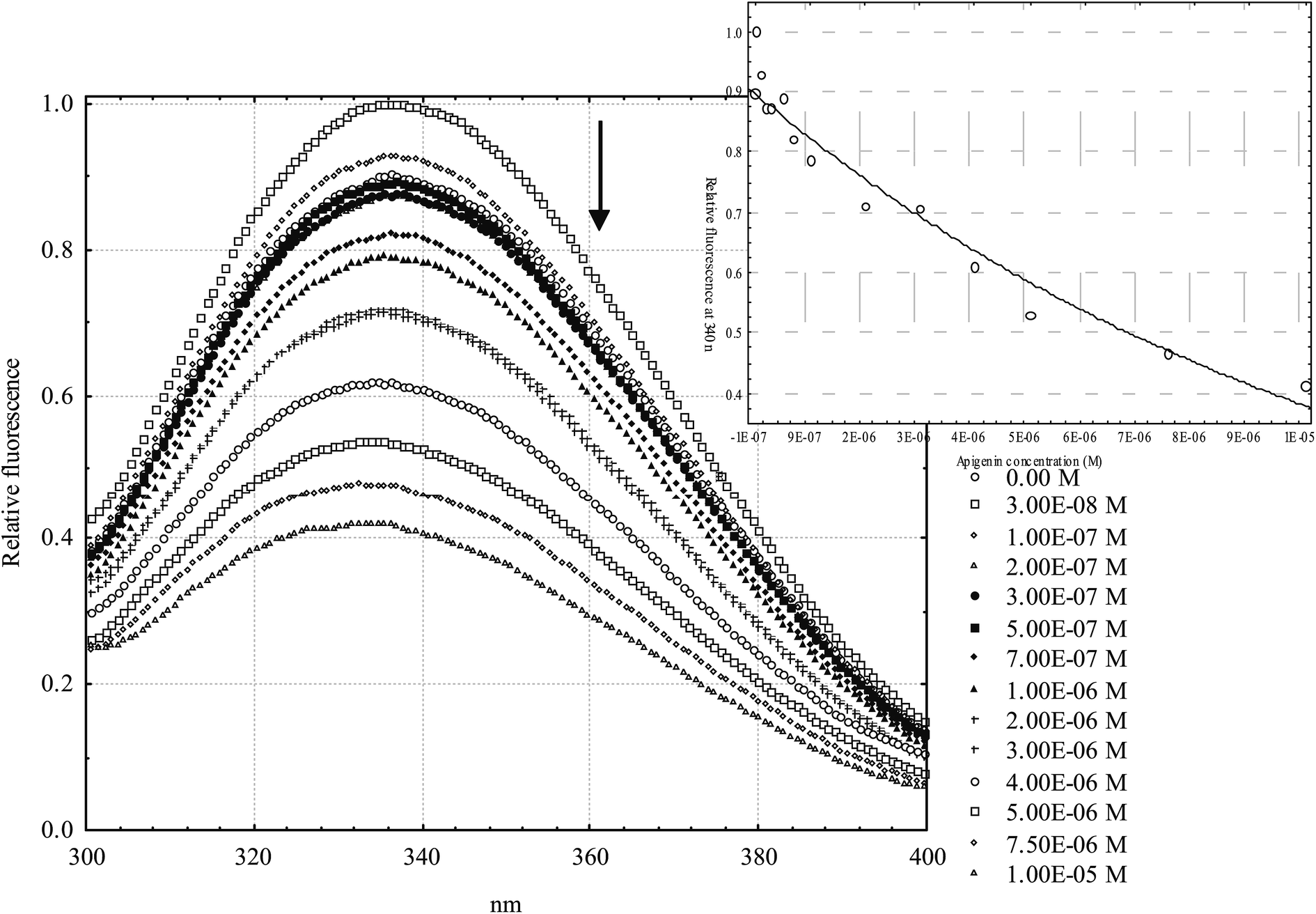 | ||
| Fig. 3 Spectrofluorimetric titration of HSA with apigenin. Total concentration of HSA was held constant at 1 μM and total concentrations of apigenin varied from 0 (top spectra) to 10 μM (bottom spectra) incubated in pH 7.4 buffer at 25 °C. Inset: fitting curve at 340 nm. | ||
3.2. DFT calculations for structure–affinity relationship of flavonoid binding to HSA
At physiological pH analyzed flavonoids have a tendency to bind to the IIA binding site of HSA in the form of an anion. There are several key flavonoid properties that are associated with respective binding affinity: (1) C3 nucleophilicity and the partial charge linked to (2) the O4 partial charge, (3) electrophilicity of C8 substituent, (4) high HOMO and LUMO energies, and (5) coplanarity of both AC and B rings and A and C rings. Very similar results for structures optimized in vacuo and in the model solvent (ε = 78.4) were obtained.According to Fig. 4, nucleophilicity and the partial charge of C3 atom enable separation of analyzed flavonoid classes which correlates with the affinity for HSA. Unfortunately these properties of the C3 atom do not enable within-group differentiation between molecules with high and low HSA affinity. It may be noticed that high C3 nucleophilicity induced by the presence of a hydroxyl group increases flavonoid affinity for HSA. Flavonols and flavones possess the highest affinity for HSA: alteration of steric properties due to substitutions at C2 and C3 atoms significantly reduces their affinity for HSA, as it is shown for isoflavones.24
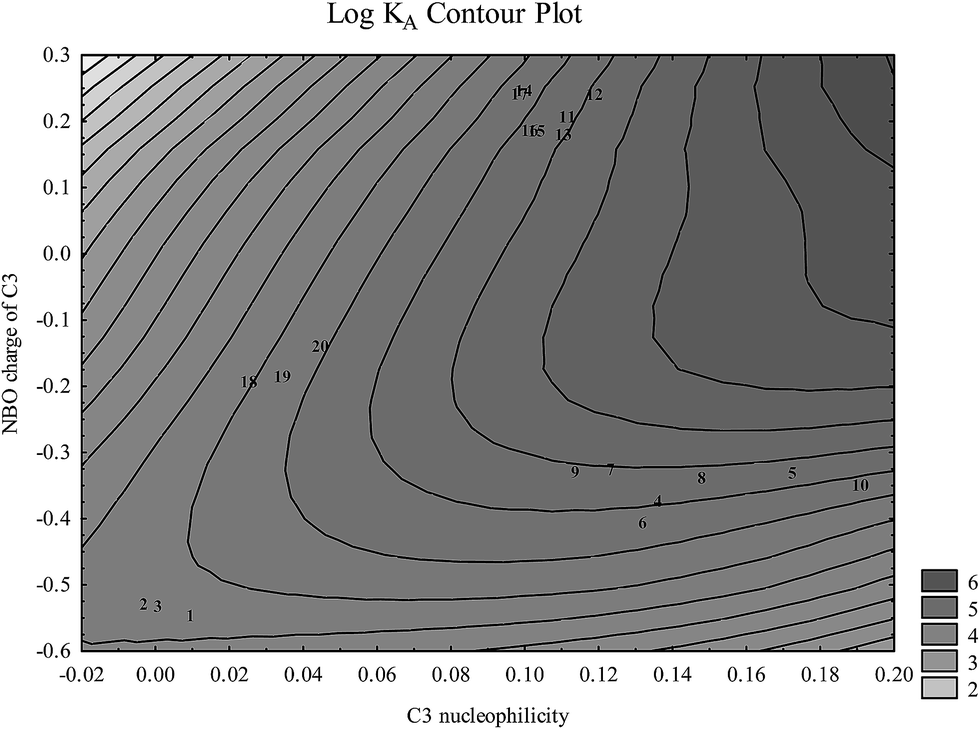 | ||
| Fig. 4 Flavonoid aglycone classes depicted in space nucleophilicity and partial charge of C3 atom calculated at the B3LYP level (numbers correspond to flavonoids in Table 1). | ||
Fig. 5 reveals strong dependence of logKA on partial charge of the O4: flavonoid affinity for HSA decreases with decreasing negative charge. This association underlines the general significance of polar interactions between O4 and HSA previously described for luteolin27 and also possible interaction with the Lys 195 residue located nearby.21 Among other types of polar interactions hydrogen bonding is as the most probable type of interaction: oxo-group attached to the C4 atom represents a good hydrogen bond acceptor that may increase the affinity for HSA.
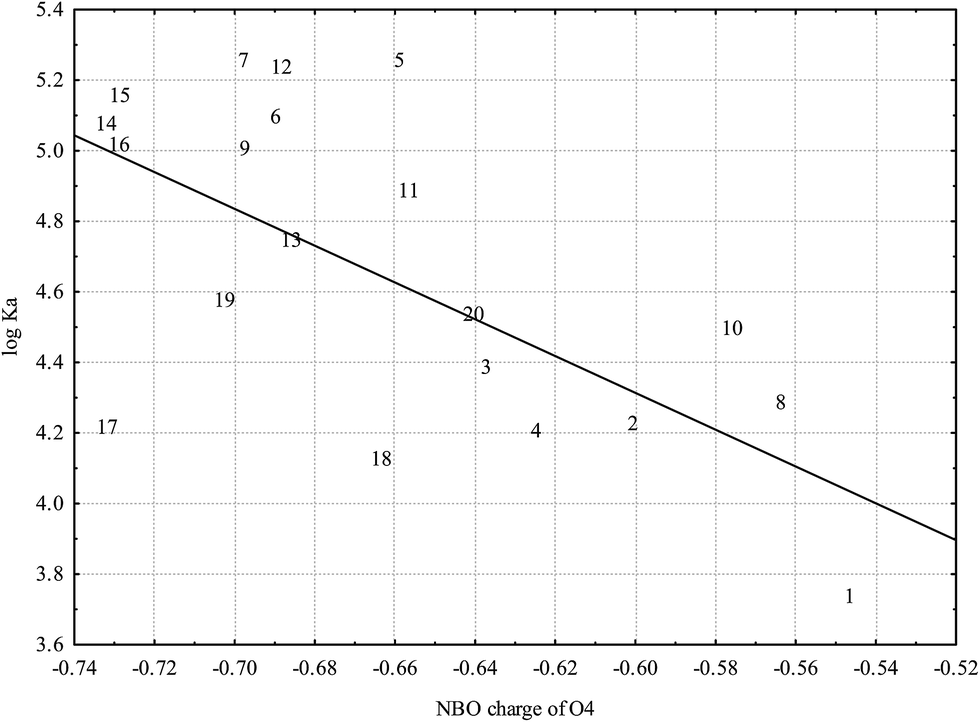 | ||
| Fig. 5 Dependence of logKA on O4 partial charge based on NBO analysis (r = −0.6514; p = 0.0019) calculated at the B3LYP level (numbers correspond to flavonoids in Table 1). | ||
Most of the previous studies emphasized the significance of nonpolar interactions of the ring A and its substituents with the HSA.25,27,75 Electronic properties of A ring atoms and substituents have been analyzed in that respect.
The only significant association between electronic properties of A ring substituents and HSA binding established is depicted in Fig. 6: very low electrophilicity of C8 substituent is associated with higher affinity for HSA. Fig. 6 suggests that low electrophilicity of the C8 substituent is relevant, but not sufficient for effective binding to HSA.
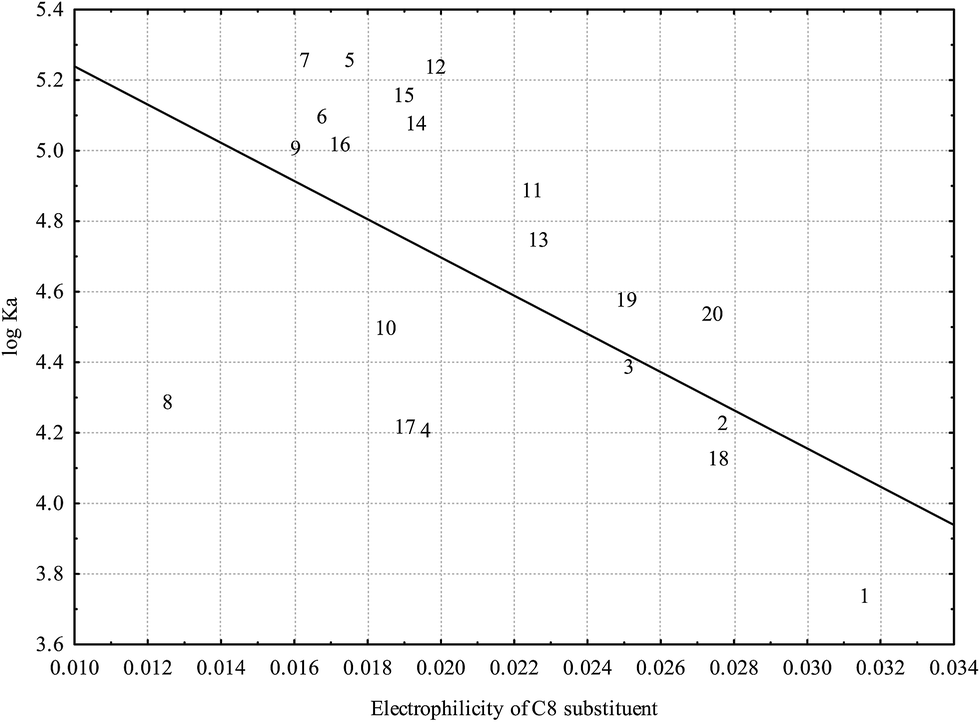 | ||
| Fig. 6 Relationship between logKA and electrophilicity of C8 substituent calculated at the B3LYP level (r = −0.5862; p = 0.0066) (numbers correspond to flavonoids in Table 1). | ||
Fig. 7 shows that tightly bound flavonoid ions are characterized by high HOMO and LUMO energies. This is consistent with their tendency towards multiple hydrogen bond formations in which flavonoid ions may play both hydrogen bond acceptor and hydrogen bond donor roles.
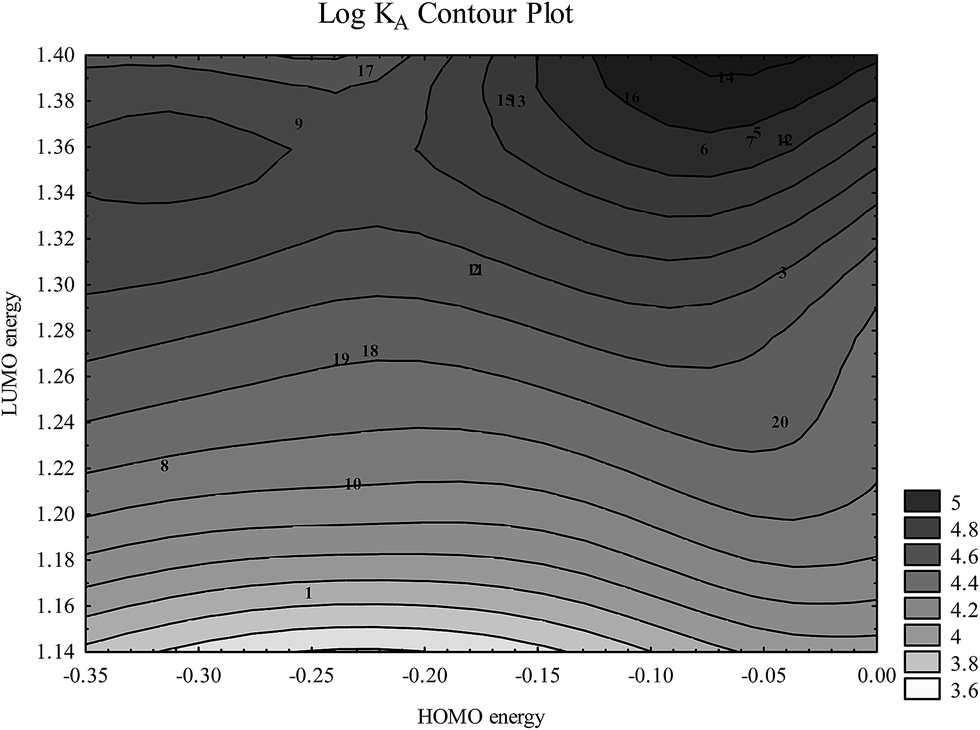 | ||
| Fig. 7 Dependence of logKA on HOMO and LUMO energies (presented in a.u.) (numbers correspond to flavonoids in Table 1). | ||
KA in both in vacuo (r = 0.0206; p = 0.9313) and the explicit solvation (r = 0.1654; p = 0.4858) shows that conjugation has no effect on HSA binding. This is in contradiction with some earlier studies,24,75 as well as with the impact of the C8 electrophilicity. Although the present study includes a large number of flavonoids, there is still a relatively low variation in energy gaps. This limits the significance of this notion strictly to the four selected classes of flavonoid aglycones.
Flavonoid aglycone geometries presented here were calculated without taking HSA into account. Obtained results are consistent with the results published by Aparicio,51 who has revealed that flavonoid aglycones can form intramolecular hydrogen bonds that stabilize coplanar geometries. It is interesting to notice that coplanarity of AC and B rings (Fig. 2) in terms of dihedral angle (angle between atoms 3-2-1′-2′ for flavanones, flavones and flavonols, and 2-3-1′-2′ for isoflavones), is associated with HSA binding affinity in vacuo (r = −0.6832; p = 0.0009), but this association is not present in the solvation water model (r = −0.0349; p = 0.8838). Considering that flavonoid geometries attached to HSA calculated in docking studies show lack of coplanarity of AC and B rings,21,25,26,28 we incline to the conclusion that coplanarity obtained in vacuo only reflects the propensity of flavonoids towards hydrogen bonding to HSA.
Also, it has been determined that AC ring planarity is necessary (dihedral angle between planes defined by atoms 4-4a-5 and 4a-5-6), but not sufficient for effective binding to HSA (r = −0.5097; p = 0.0217 and r = −0.5093; p = 0.0218 for in vacuo and explicit solvation, respectively): even a small deviation from planarity is associated with the loss of affinity for HSA, but all planar molecules do not bind to HSA with equal affinity. Since planarity is frequently associated with conjugation some authors expected that flavonoids tightly bound to HSA establish respective interaction through conjugation.24,75 According to our results, it seems that the planarity of flavonoids itself makes greater impact on HSA binding than conjugation: flavanones which are characterized by significantly lower binding constants than other flavonoids speak in favor of this notion. Moreover, according to the literature, it has been established that the primary binding site for flavanones is not, as opposed to other flavonoids, in subdomain IIA, but is located closer to the binding site in the IIIA subdomain.23 Possible reason for this effect could be the presence of a non-planar C ring, which disables binding in the vicinity of Trp 214 residue of the IIA subdomain.
4. Conclusions
In this study spectroscopically determined binding affinities of flavonoid aglycones for HSA were associated with their steric and electronic features. Nucleophilicity and partial charge of the C3 atom enabled classification of flavonoids into subgroups: flavanones (very low nucleophilicity and high negative partial charge), isoflavones (low nucleophilicity and low negative partial charge), flavones (medium to high nucleophilicity and medium negative partial charge) and flavonols (medium nucleophilicity and positive partial charge), with flavones and flavonols being most tightly bound. Increased negative partial charge of the O4 atom has shown strong association with the HSA binding affinity, reflecting its good hydrogen acceptor properties. Also, it has been shown that planarity is of great importance for the flavonoid binding. Coplanarity of A and C rings is associated with higher binding constants. According to our calculations it is a prerequisite for binding in the hydrophobic cavity. Additionally, coplanarity of AC and B rings reflects the propensity of flavonoids towards hydrogen bonding to HSA, consistent with corresponding high HOMO and LUMO energies. Contrary to previously published results, this study shows that neither conjugation of B and AC rings nor hydrogen acceptor and donor properties of the B ring are common determinants of flavonoid binding to HSA; however a negative charge located on the B ring shows a favorable effect on the binding constant.This study represents the most extensive study of flavonoids binding to HSA, complementing an experimental technique of fluorescence spectrophotometry with results of quantum chemical approach to provide explanations of flavonoid binding properties. However, it is important to emphasize that all of the laboratory measurements were done in vitro. Further studies are needed to evaluate biological implications of the described phenomenon like flavonoid–drug and flavonoid–hormone interactions.
Acknowledgements
We thank Dr Sc. Kevin M. Johnson for proofreading the manuscript. This work has been fully supported by Croatian Science Foundation under the project UIP-2014-09-5704.References
- T. Peters, in All about Albumin, Academic Press, San Diego, 1996, pp. 251–284 Search PubMed.
- X. M. He and D. C. Carter, Nature, 1992, 358, 209–215 CrossRef CAS PubMed.
- T. Peters, in All about Albumin, Academic Press, San Diego, 1996, pp. 76–132 Search PubMed.
- G. Sudlow, D. J. Birkett and D. N. Wade, Mol. Pharmacol., 1975, 11, 824–832 CAS.
- G. Sudlow, D. J. Birkett and D. N. Wade, Mol. Pharmacol., 1976, 12, 1052–1061 CAS.
- J. Ghuman, P. A. Zunszain, I. Petitpas, A. A. Bhattacharya, M. Otagiri and S. Curry, J. Mol. Biol., 2005, 353, 38–52 CrossRef CAS PubMed.
- C. E. Petersen, C.-E. Ha, S. Curry and N. V. Bhagavan, Proteins, 2002, 47, 116–125 CrossRef CAS PubMed.
- M. R. Housaindokht, Z. Rouhbakhsh Zaeri, M. Bahrololoom, J. Chamani and M. R. Bozorgmehr, Spectrochim. Acta, Part A, 2012, 85, 79–84 CrossRef CAS PubMed.
- A. Dasgupta, Clin. Chim. Acta, 2007, 377, 1–13 CrossRef CAS PubMed.
- B. Leboulanger, R. H. Guy and M. B. Delgado-Charro, Eur. J. Pharm. Sci., 2004, 22, 427–433 CrossRef CAS PubMed.
- U. Kragh-Hansen, Pharmacol. Rev., 1981, 33, 17–53 CAS.
- K. Yamasaki, V. T. G. Chuang, T. Maruyama and M. Otagiri, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 5435–5443 CrossRef CAS PubMed.
- T. J. Mabry, K. R. Markham and M. B. Thomas, The Systematic Identification of Flavonoids, Springer-Verlag, Berlin, Heidelberg, New York, 1970 Search PubMed.
- W. Bors, C. Michel and K. Stettmaier, BioFactors, 1997, 6, 399–402 CrossRef CAS PubMed.
- M. Foti, M. Piattelli, M. T. Baratta and G. Ruberto, J. Agric. Food Chem., 1996, 44, 497–501 CrossRef CAS.
- L. Dauchet, L. Dauchet, P. Amouyel, P. Amouyel, S. Hercberg, S. Hercberg, J. Dallongeville and J. Dallongeville, J. Nutr., 2006, 136, 2588–2593 CAS.
- I. C. W. Arts and P. C. H. Hollman, Am. J. Clin. Nutr., 2005, 81, 317S–325S CAS.
- M. Bojić, Ž. Debeljak, M. Medić-Šarić and M. Tomičić, Clin. Chem. Lab. Med., 2012, 50, 1403–1408 CrossRef PubMed.
- C. Dufour and O. Dangles, Biochim. Biophys. Acta, Gen. Subj., 2005, 1721, 164–173 CrossRef CAS PubMed.
- S. Bi, L. Ding, Y. Tian, D. Song, X. Zhou, X. Liu and H. Zhang, J. Mol. Struct., 2004, 703, 37–45 CrossRef CAS.
- F. Zsila, Z. Bikádi, M. Simonyi and Z. Bika, Biochem. Pharmacol., 2003, 65, 447–456 CrossRef CAS PubMed.
- C. D. Kanakis, P. A. Tarantilis, M. G. Polissiou, S. Diamantoglou and H. A. Tajmir-Riahi, J. Mol. Struct., 2006, 798, 69–74 CrossRef CAS.
- M. K. Khan, N. Rakotomanomana, C. Dufour and O. Dangles, Food Funct., 2011, 2, 617 CAS.
- M. Poór, S. Kunsági-Máté, T. Bencsik, J. Petrik, S. Vladimir-Knežević and T. Koszegi, Int. J. Biol. Macromol., 2012, 51, 279–293 CrossRef PubMed.
- H. G. Mahesha, S. A. Singh, N. Srinivasan and A. G. Appu Rao, FEBS J., 2006, 273, 451–467 CrossRef CAS PubMed.
- Y. Li, W. He, H. Liu, X. Yao and Z. Hu, J. Mol. Struct., 2007, 831, 144–150 CrossRef CAS.
- Z. Jurasekova, G. Marconi, S. Sanchez-Cortes and A. Torreggiani, Biopolymers, 2009, 91, 917–927 CrossRef CAS PubMed.
- S. R. Feroz, S. B. Mohamad, Z. S. D. Bakri, S. N. A. Malek and S. Tayyab, PLoS One, 2013, 8, e76067 CAS.
- J. Dai, T. Zou, L. Wang, Y. Zhang and Y. Liu, Luminescence, 2014, 29, 1154–1161 CrossRef CAS PubMed.
- C.-Z. Lin, M. Hu, A.-Z. Wu and C.-C. Zhu, J. Pharm. Anal., 2014, 4, 392–398 CrossRef CAS.
- M. R. Eftink and C. A. Ghiron, Anal. Biochem., 1981, 114, 199–227 CrossRef CAS PubMed.
- T. Peters, in All about Albumin, Academic Press, San Diego, 1996, pp. 9–75 Search PubMed.
- O. Dangles, C. Dufour and S. Bret, J. Chem. Soc., Perkin Trans. 2, 1999, 737–744 RSC.
- B. Sengupta and P. K. Sengupta, Biochem. Biophys. Res. Commun., 2002, 299, 400–403 CrossRef CAS PubMed.
- R. S. Keri, S. Budagumpi, R. K. Pai and R. G. Balakrishna, Eur. J. Med. Chem., 2014, 78, 340–374 CrossRef CAS PubMed.
- M. Singh, M. Kaur and O. Silakari, Eur. J. Med. Chem., 2014, 84, 206–239 CrossRef CAS PubMed.
- R. Dulbecco and M. Vogt, J. Exp. Med., 1954, 99, 167–182 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- H. Gampp, M. Maeder, C. J. Meyer and A. D. Zuberbühler, Talanta, 1985, 32, 95–101 CrossRef CAS PubMed.
- H. Gampp, M. Maeder, C. J. Meyer and A. D. Zuberbühler, Talanta, 1985, 32, 257–264 CrossRef CAS PubMed.
- H. Gampp, M. Maeder, C. J. Meyer and A. D. Zuberbühler, Talanta, 1986, 33, 943–951 CrossRef CAS PubMed.
- M. Maeder and Y.-M. Neuhold, in Practical Data Analysis in Chemistry, 2007, vol. 10 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., 2009.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- T. U. Rahman, M. Arfan, T. Mahmood, W. Liaqat, M. A. Gilani, G. Uddin, R. Ludwig, K. Zaman, M. I. Choudhary, K. F. Khattak and K. Ayub, Spectrochim. Acta, Part A, 2015, 146, 24–32 CrossRef PubMed.
- E. Klein, J. Rimarčík, E. Senajová, A. Vagánek and J. Lengyel, Comput. Theor. Chem., 2016, 1085, 7–17 CrossRef CAS.
- K. S. Lau, A. Mantas, G. A. Chass, F. H. Ferretti, M. Estrada, G. Zamarbide and I. G. Csizmadia, Can. J. Chem., 2002, 80, 845–855 CrossRef CAS.
- S. Aparicio, Int. J. Mol. Sci., 2010, 11, 2017–2038 CrossRef CAS PubMed.
- K. S. P. Perry, T. J. Nagem and W. B. De Almeida, Struct. Chem., 1999, 10, 277–284 CrossRef CAS.
- S. Abbate, L. F. Burgi, E. Castiglioni, F. Lebon, G. Longhi, E. Toscano and S. Caccamese, Chirality, 2009, 21, 436–441 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- J. R. Pliego, Chem. Phys., 2004, 306, 273–280 CrossRef CAS.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1 Search PubMed.
- P. Bultinck, R. Carbó-Dorca and W. Langenaeker, J. Chem. Phys., 2003, 118, 4349–4356 CrossRef CAS.
- P. Bultinck, S. Van Damme and A. Cedillo, J. Comput. Chem., 2013, 34, 2421–2429 CrossRef CAS PubMed.
- J. Melin, P. W. Ayers and J. V. Ortiz, J. Phys. Chem. A, 2007, 111, 10017–10019 CrossRef CAS PubMed.
- K. Yamasaki, T. Maruyama, A. Takadate, A. Suenaga, U. Kragh-Hansen and M. Otagiri, J. Pharm. Sci., 2004, 93, 3004–3012 CrossRef CAS PubMed.
- A. Banerjee, K. Basu and P. K. Sengupta, J. Photochem. Photobiol., B, 2008, 90, 33–40 CrossRef CAS PubMed.
- J. Xiao, H. Cao, Y. Wang, K. Yamamoto and X. Wei, Mol. Nutr. Food Res., 2010, 54(suppl. 2), S253–S260 CAS.
- J.-L. Yuan, Z. Iv, Z.-G. Liu, Z. Hu and G.-L. Zou, J. Photochem. Photobiol., A, 2007, 191, 104–113 CrossRef CAS.
- M. Poór, Y. Li, S. Kunsági-Máté, J. Petrik, S. Vladimir-Knežević and T. Koszegi, J. Lumin., 2013, 142, 122–127 CrossRef.
- B. Tu, Z.-F. Chen, Z.-J. Liu, R.-R. Li, Y. Ouyang and Y.-J. Hu, RSC Adv., 2015, 5, 73290–73300 RSC.
- G. Zhang, L. Wang and J. Pan, J. Agric. Food Chem., 2012, 60, 2721–2729 CrossRef CAS PubMed.
- M. Voicescu and R. Bandula, Spectrochim. Acta, Part A, 2015, 138, 628–636 CrossRef CAS PubMed.
- J. Ma, Y. Liu, L. Chen, Y. Xie, L. Y. Wang and M. X. Xie, Food Chem., 2012, 132, 663–670 CrossRef CAS PubMed.
- I. Matei, S. Ionescu and M. Hillebrand, J. Lumin., 2011, 131, 1629–1635 CrossRef CAS.
- O. Dangles, C. Dufour, C. Manach, C. Mornad and C. Remesy, Methods Enzymol., 2001, 335, 319–333 CAS.
- A. Bolli, M. Marino, G. Rimbach, G. Fanali, M. Fasano and P. Ascenzi, Biochem. Biophys. Res. Commun., 2010, 398, 444–449 CrossRef CAS PubMed.
- B. Mishra, A. Barik, K. I. Priyadarsini and H. Mohan, J. Chem. Sci., 2005, 117, 641–647 CrossRef CAS.
- Y. Li, W. He, Y. Dong, F. Sheng and Z. Hu, Bioorg. Med. Chem., 2006, 14, 1431–1436 CrossRef CAS PubMed.
- Q. Bian, J. Liu, J. Tian and Z. Hu, Int. J. Biol. Macromol., 2004, 34, 275–279 CrossRef PubMed.
- S. Pal and C. Saha, J. Biomol. Struct. Dyn., 2013, 32, 1132–1147 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |