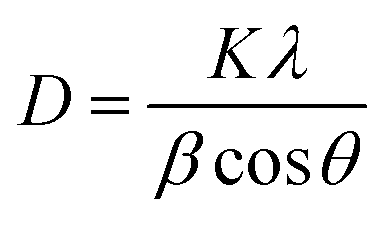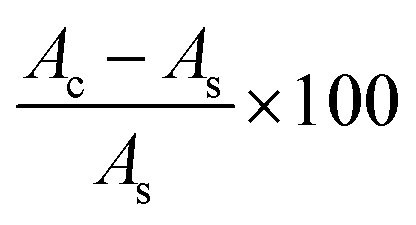DOI:
10.1039/C6RA17569D
(Paper)
RSC Adv., 2016,
6, 81436-81446
Antimicrobial, antioxidant and anticancer activity of biogenic silver nanoparticles – an experimental report†
Received
9th July 2016
, Accepted 14th August 2016
First published on 15th August 2016
Abstract
In the present study, use of a Helicteres isora stem bark extract for the biosynthesis of AgNPs is described. It was observed that the aqueous silver (Ag+) ions, once associated in the stem bark extract, were reduced in solution, thereby leading to formation of the stable AgNPs. These AgNPs were characterized using several techniques. The nanoparticles show a maximum absorbance at 419–431 nm in the ultraviolet-visible spectra. The presence of the steroid sapogenin was identified using Fourier transform-infrared (FTIR) spectroscopy. The reduction of the Ag+ ions to elemental silver was investigated by using X-ray diffraction (XRD). Transmission electron microscopy (TEM) revealed the formation of monodisperse, with low polydispersity, nanoparticles of 25.55 nm, and the presence of elemental silver was confirmed through energy dispersive spectroscopy (EDX) analysis. The AgNPs showed antioxidant activities such as DPPH, hydrogen peroxide and nitric oxide radical scavenging and a reducing power compared to the standard compounds. The antibacterial effect was determined against test strains, showing significant inhibition. The antiproliferative activity of the AgNPs was demonstrated using oral carcinoma (KB) cells with MTT, and confirmed using AO/EtBr, comet assay, DCFH-DA and Rhodamine 123 staining. In the toxicity study, a significant mortality rate was observed against Artemia with an IC50 concentration of 70 μg mL−1 and 108 h exposure. The NPs showed as cytotoxic against Artemia at 108 h, so they are cytotoxic at high concentrations and after prolonged exposure.
1. Introduction
Worldwide cancer is the leading cause of death. In India about 6% of deaths are due to cancer, which contributes 8% of the global cancer mortality. Oral cancer is in India the third and worldwide the eleventh most common cancer with over 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 new cases annually, and over 30% of all cancers and 90% of oral cancers have been caused by tobacco use and excessive alcohol consumption.1 All of the tobacco consumption developed oral cancers have annual incidence rates ranging from 5.2/1000 to 30.2/1000, whereas non-users develop the fewest oral lesions with incidence rates from 0.6/1000. An increasing number of teenagers are being affected and 25% of the cases have no associated risk factors.2 For the treatment of oral cancer, some possible methods are surgery, radiation therapy, chemotherapy, targeted therapy and palliative treatment but the cost is high. So, in the 21st century, nanotechnology is an emerging technology and it has incredible applications in medicine, drug delivery,3 molecular imaging,4 wound healing,5 the diagnosis of cardiovascular diseases,6 antibacterial processes,7,8 photocatalytic degradation9,10 and amperometry.11 The major interest has been the development of new particles and investigation of their properties by fine-tuning the particle shape, size and distribution.
000 new cases annually, and over 30% of all cancers and 90% of oral cancers have been caused by tobacco use and excessive alcohol consumption.1 All of the tobacco consumption developed oral cancers have annual incidence rates ranging from 5.2/1000 to 30.2/1000, whereas non-users develop the fewest oral lesions with incidence rates from 0.6/1000. An increasing number of teenagers are being affected and 25% of the cases have no associated risk factors.2 For the treatment of oral cancer, some possible methods are surgery, radiation therapy, chemotherapy, targeted therapy and palliative treatment but the cost is high. So, in the 21st century, nanotechnology is an emerging technology and it has incredible applications in medicine, drug delivery,3 molecular imaging,4 wound healing,5 the diagnosis of cardiovascular diseases,6 antibacterial processes,7,8 photocatalytic degradation9,10 and amperometry.11 The major interest has been the development of new particles and investigation of their properties by fine-tuning the particle shape, size and distribution.
Green nanotechnology is a developing area due to the removal of harmful reagents and it provides an effective synthesis of expected products at a low cost. Noble metal particles exhibit new physicochemical properties that are not observed either in bulk metals or individual molecules. Among the noble metal nanoparticles, silver nanoparticles (AgNPs) in particular are known for their versatile applications in medical industries and defense,12,13 and they are applied spectrally for active camouflage in the military.14 Also, AgNPs were the most efficient as antimicrobials and antioxidants.8,15 Plant extracts have less toxic substances and require minimal purification steps when compared to chemical methods.16 AgNPs have been studied for their capability to diminish microbial infections of the skin and also to prevent bacterial colonization.17 There are several physical and chemical methods employed for the synthesis of AgNPs,18 but these methods have created great environmental issues with the use of toxic substances and the generation of harmful by-products. But, the use of natural entities like microorganisms and plant extracts has been explored for the synthesis of AgNPs and this has been found to be a clean, nontoxic, low cost and eco-friendly method. While using microorganisms has some disadvantages, which include maintaining the cell culture in an aseptic environment, plant mediated synthesis offers several advantages such as a low cost and nontoxic and eco-friendly by-products.19 Plant extracts act as reducing and capping mediators in the synthesis of AgNPs and the particles formed are more stable with different shapes and dimensions, making this a more efficient method than other synthesis methods. Recently, many medicinal plants have been employed for the synthesis of AgNPs such as Cleistanthus collinus,15 Morinda citrifolia,20 Alternanthera sessilis21 and Ceropegia thwaitesii.8
In this paper, we will concentrate our attention on the green synthesis of AgNPs using a Helicteres isora stem bark extract and analysis of their antioxidant and antibacterial activities against human pathogens. Furthermore, the anticancer activity was also determined by investigating their effect on cancer cell viability and measuring the intracellular ROS, apoptotic morphological changes, mitochondrial membrane potential (MMP) changes and DNA damage in oral carcinoma (KB) cells. Also, the present study aims at examining whether AgNPs are toxic to marine brine shrimp, using Artemia salina as a toxicology model organism, and whether the toxicity of AgNPs is related to release of Ag+ from the AgNPs or to specific effects of the AgNPs. The toxic effects of AgNPs on the mortality of Artemia salina are assessed and discussed in the present study.
2. Results and discussion
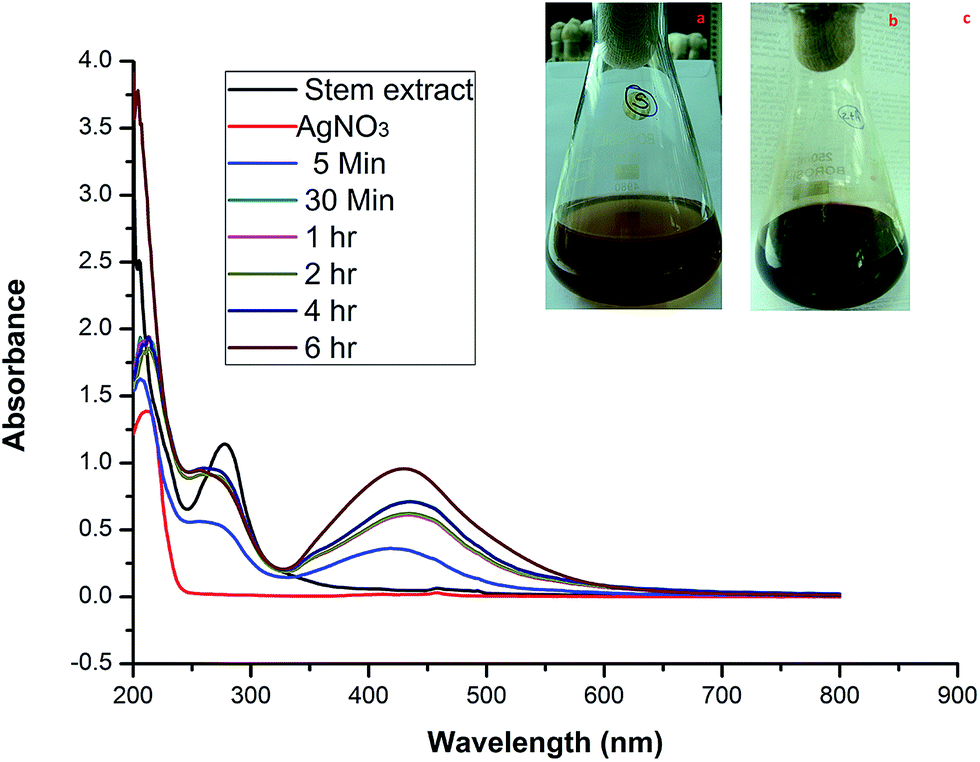
Generally NPs are categorized by their shape, size, surface area, and dispersity. In many applications, these properties of homogeneity are playing a key role.22 When the stem bark extract was combined with AgNO3 and allowed to react at room temperature, within 30 min of reaction the color changed from brown to dark brown (Fig. 1a–c), indicating the formation of AgNPs. It is an efficient and rapid method, which has been very well explained by some investigators who have worked with different plant systems.8,15 During the reaction, the color change was due to the excitation of surface plasmon vibrations in the metal nanoparticles.23 Our results are in agreement with previous report,8 in which the formation of AgNPs within 30 min of incubation is reported.
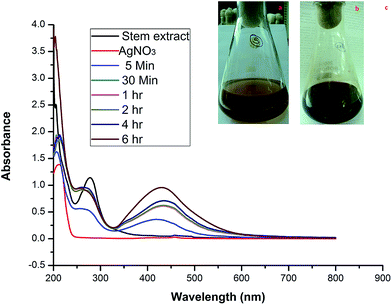 |
| | Fig. 1 Photographs of (a) the extract and (b) the color change after adding AgNO3. (c) UV-vis spectra of the AgNPs synthesized using stem bark extract after different incubation times. | |
2.1. Characterization
2.1.1. UV-visible spectra. In order to verify the synthesis of AgNPs, the samples were subjected to UV-vis analysis. Observation revealed that the reduction of the Ag+ to Ag0 during the exposure to the extract filtrate can be easily followed visually and using UV-vis spectroscopy (Fig. 1a and b). This analysis showed a sharp absorbance at 430 nm (Fig. 1c). The UV-vis absorption band in the visible light region (420–450 nm) is evidence of the presence of surface plasmon resonance (SPR) of the AgNPs.8,24 The formation of SPR peaks depends on various factors like particle formation, size and shape.25,26 Two or additional SPR bands correspond to anisotropic molecules, whereas a single SPR band represents the spherical form of nanoparticles.27 Sands28 stated that the presence of two SPR bands for the reaction mixture reveals a cubic nature (with Oh symmetry) of the biosynthesized AgNPs. So, the current biosynthesized AgNPs are of a cubic nature because double SPR bands were exhibited in the spectrum. The SPR peak intensity increased with the reaction time showing an increasing concentration of AgNPs.29
2.1.2. DLS. DLS was used to analyze quantitatively the size distributions and monodispersity in the colloidal suspensions. The DLS study revealed the differential intensity, number and zeta potential related to the particle size distributions of the biosynthesized AgNPs (ESI Fig. S1a–c†). The average particle size was found to be 64.5 nm is calculated by DLS. The zeta potential of the colloidal solution was found to be −19.1 mV. The size of the particles obtained was much greater compared to the TEM analysis and XRD results. This polydispersity is due to the fact that the measured size also included the biomaterials covering the surface of silver nanoparticles.30
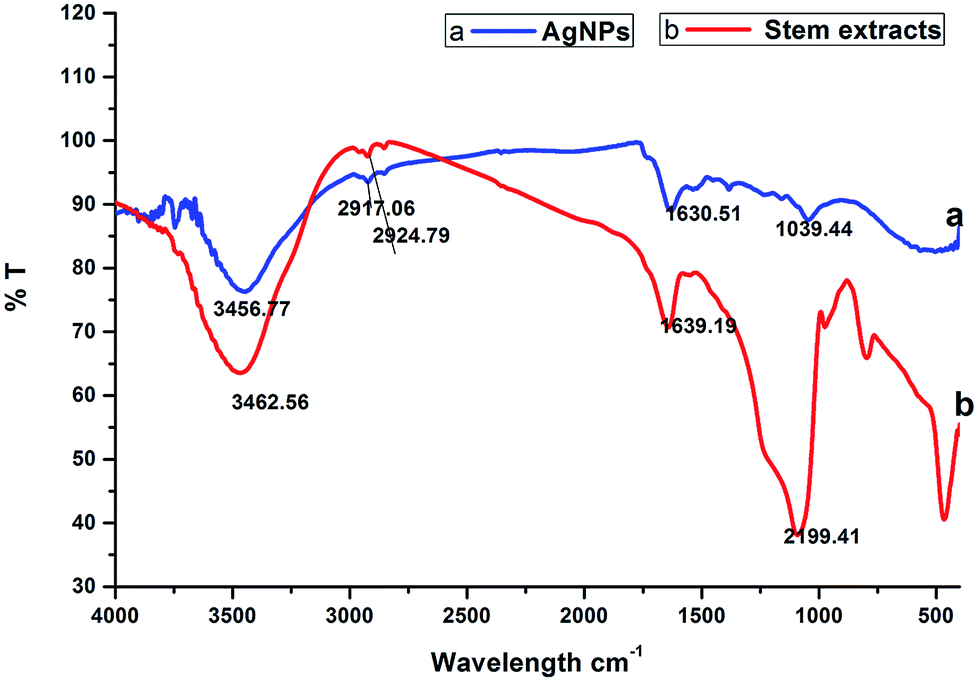
2.1.3. FTIR spectral analysis. Fig. 2 shows the FTIR spectrum of the synthesized AgNPs, which reveals the probable biomolecules existing in the stem bark extract that are accountable for the reduction of the silver ions and its interaction with the AgNPs. The IR spectrum of the AgNPs shows intense bands at 3456.77 cm−1, 2917.06 cm−1, 1630.51 cm−1 and 1039.44 cm−1 (Fig. 2a). The IR spectrum of the extract shows intense bands at 3462.56 cm−1, 2924.79 cm−1, 1639.19 cm−1 and 2199.41 cm−1 (Fig. 2b), and significant variation was observed among the spectral positions of the IR bands in the stem/bark extract and the biosynthesized AgNPs owing to the reduction process. The broad band at 3456.77 cm−1 corresponds to the strong stretching vibrations of the hydroxyl group (–OH) of the steroid sapogenin, this broad band was shifted from its position at 3462.56 cm−1 for the extract. The two sharp intense peaks at 2917.06 cm−1 and 1630.51 cm−1 can be attributed to C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations, which indicates the presence of alcohol and alkene groups in the extract that may be involved in the reduction process.31 The IR spectrum of the stem/bark extract exhibits a strong band at 1639.19 cm−1 corresponding to a CC (symmetry reduces intensity and NH2 scissoring (1° amines)) stretching mode and this peak is shifted to 1630.51 cm−1, suggesting possible association of the above mentioned groups with the AgNPs synthesis. This amine band indicates that the steroid sapogenin can bind to Ag+ through alcohol and alkene groups.32 This result suggests that the steroid sapogenin is interacting with the AgNPs and also that the secondary structure was not changed during the reaction with Ag+ ions or after binding to the biosynthesized silver nanoparticles.
C stretching vibrations, which indicates the presence of alcohol and alkene groups in the extract that may be involved in the reduction process.31 The IR spectrum of the stem/bark extract exhibits a strong band at 1639.19 cm−1 corresponding to a CC (symmetry reduces intensity and NH2 scissoring (1° amines)) stretching mode and this peak is shifted to 1630.51 cm−1, suggesting possible association of the above mentioned groups with the AgNPs synthesis. This amine band indicates that the steroid sapogenin can bind to Ag+ through alcohol and alkene groups.32 This result suggests that the steroid sapogenin is interacting with the AgNPs and also that the secondary structure was not changed during the reaction with Ag+ ions or after binding to the biosynthesized silver nanoparticles.
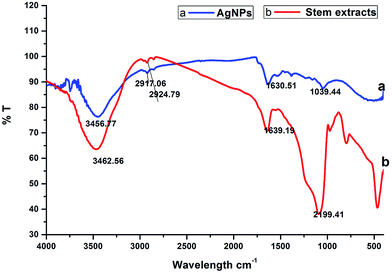 |
| | Fig. 2 FTIR analysis: (a) biosynthesized silver nanoparticles; (b) stem bark extract. | |
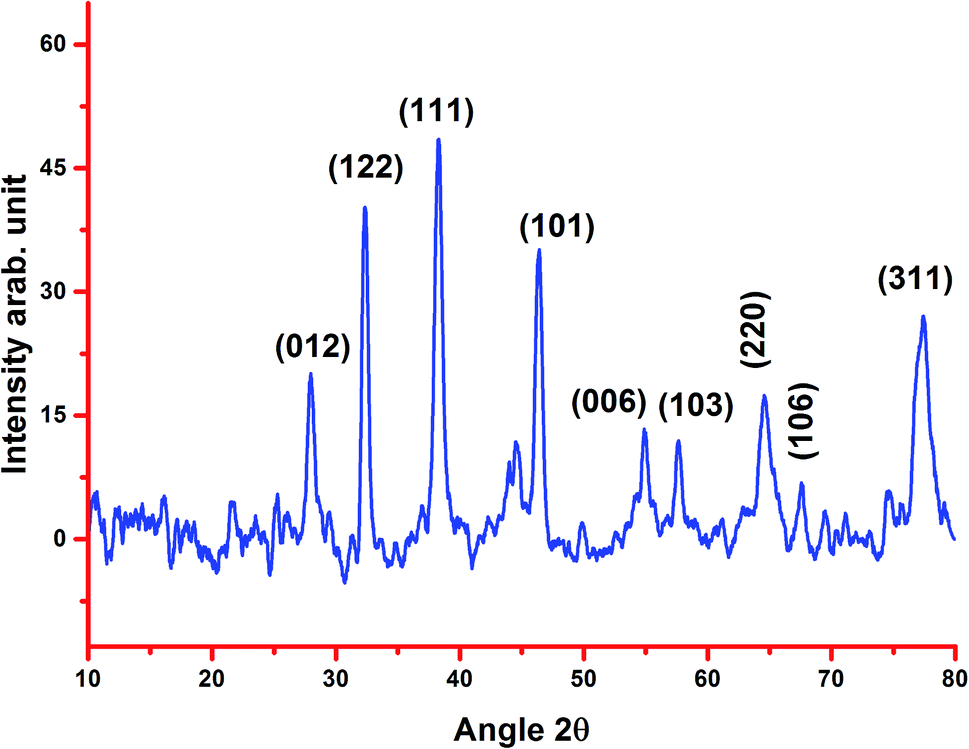
2.1.4. XRD. Fig. 3 shows the X-ray diffraction pattern of the biosynthesized AgNPs from the stem bark extract. The different distinct peaks at 27.88°, 32.41°, 38.20°, 46.37°, 55.05°, 57.90°, 64.42°, 67.49° and 77.40° indicate the (0 1 2), (1 2 2), (1 1 1), (1 0 1), (0 0 6), (1 0 3), (2 2 0), (1 0 6) and (3 1 1) reflections of silver. The XRD pattern also represents the face-centered cubic structure of silver. A sharp and strong diffraction peak positioned at 38.20° appeared, which can be assigned to the (1 1 1) reflection and closely correlates to the reported reference values of the Joint Committee on Power Diffraction Standards (JCPDS PDF no: 89-0713). The cubic crystalline nature of the biosynthesized nanoparticles was clearly indicated by the sharp peaks, which are in agreement with the previous reports.8,24,33 The average size of the crystalline AgNPs estimated using the Debye–Scherrer formula was 20.24 nm, which is less than the average size from the TEM analysis.
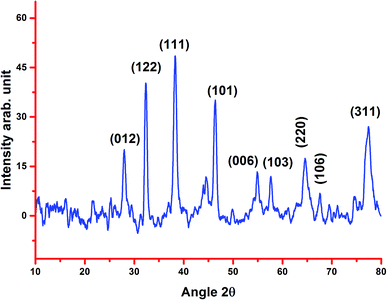 |
| | Fig. 3 XRD pattern of the biosynthesized silver nanoparticles using stem bark extract. | |
2.1.5. EDX analysis. The occurrence of elemental silver is often detected using EDX analysis (ESI Fig. S2†), which showed reduction of the silver ions to elemental silver within the reaction mixture. The EDX spectrum illustrated the presence of strong metallic Ag signals. It demonstrated elemental constituents of silver (89.75%), chlorine (6.57%) and carbon (3.68%) respectively. The most dominant sharp signal was observed at ∼3 keV for silver, which is characteristic of the absorption of crystalline AgNPs.8
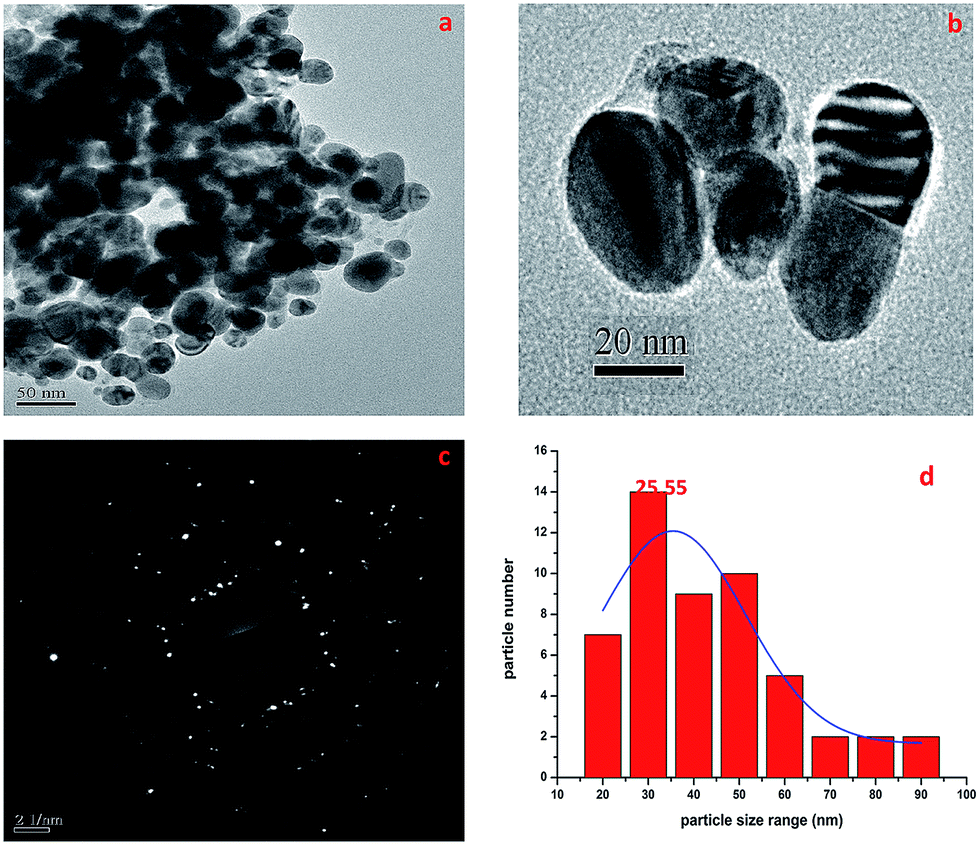
2.1.6. TEM-SAED study. The TEM micrographs of the biosynthesized AgNPs are shown in Fig. 4. It was established that the AgNPs were spherical in outline with most particles in the size range of 16–95 nm (Fig. 4d). Mostly the particles were spherical or oval in shape and aggregated, and some more individual particles were also present. The TEM image in Fig. 4b shows the biomolecular coating on the surface layer of the AgNPs, which is responsible for the enhanced stability of the biosynthesized AgNPs. The SAED pattern is shown in Fig. 4c and it confirmed the presence of elemental AgNPs, and it was also observed that the AgNPs were monodisperse with a low polydispersity index (PDI). The average crystallite size of the silver nanoparticles was estimated to be 25.55 nm (Fig. 4d).
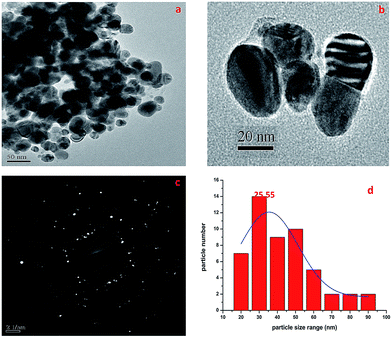 |
| | Fig. 4 TEM micrographs showing the size of the AgNPs with a SAED pattern. | |
2.2. Antioxidant activity
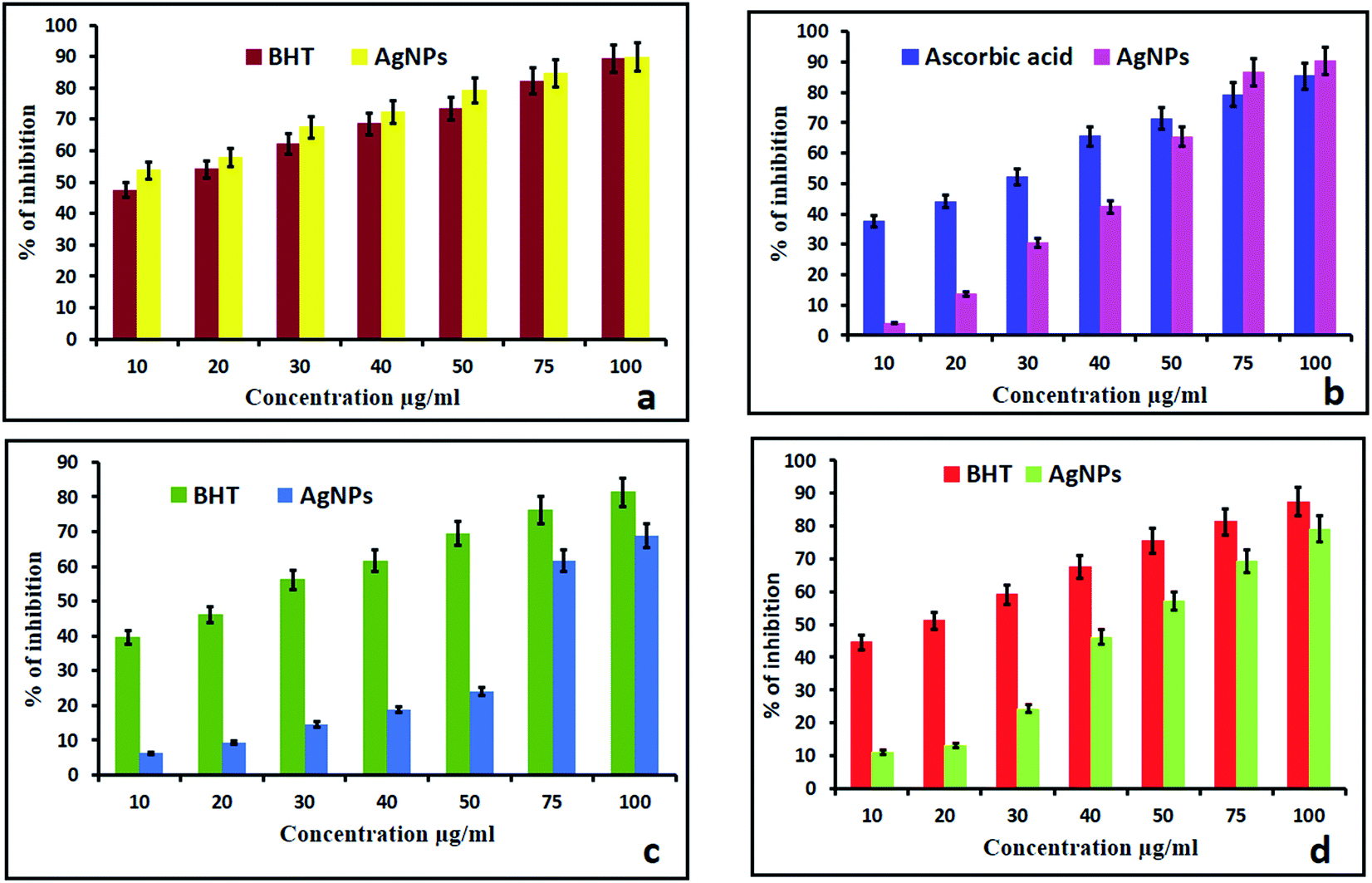
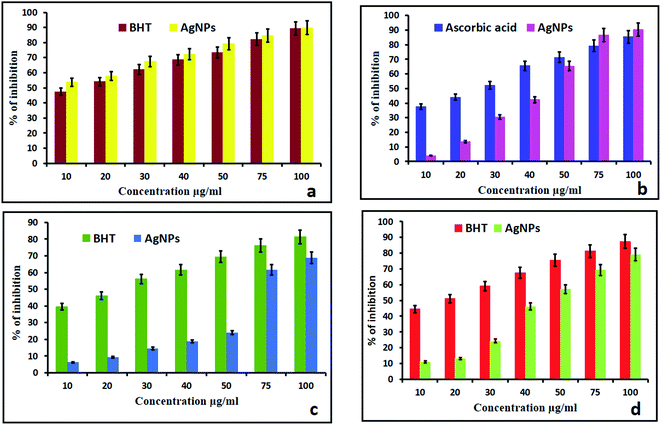
2.2.1. DPPH. DPPH is a well-known and more stable free radical for assessing the antioxidant properties of compounds. This is based on the reduction process of accepting an electron or hydrogen. The reducing ability toward DPPH was assessed spectrophotometrically by observing the color change. In the present study, the synthesized AgNPs showed better activity compared with the standard BHT. At concentrations of 10–100 μg mL−1, the AgNPs showed scavenging rates ranging from 49.89% to 91%. The above observed results were higher than those of the standard BHT (89.35%). The methanolic solution of DPPH turned from blue to light yellow with the addition of the AgNPs, which is due to the scavenging of DPPH due to donation of hydrogen molecules to stabilize the DPPH molecules.15,34 As the concentration of the AgNPs increased from 10 μg mL−1 to 100 μg mL−1, the absorbance at 517 nm decreased, thus indicating an increase in the free radical scavenging activity of the biosynthesized AgNPs (Fig. 5a).
 |
| | Fig. 5 Antioxidant activity of the biosynthesized AgNPs. | |
2.2.2. H2O2 scavenging assay. The effect of the AgNPs on the scavenging of H2O2 free radicals is shown in Fig. 5b. The inhibition at a concentration of 100 μg mL−1 was found to be 93.31% and 89.35% for the AgNPs and ascorbic acid respectively. The above observed H2O2 free radical activity was higher than that of the standard ascorbic acid due to the size and crystalline nature of the AgNPs. The dispersed AgNPs can encourage reactive oxygen species like hydroxyl radicals to form in the presence of hydrogen peroxide. H2O2 inside a cell in small amounts has been reported to accelerate the dissolution of AgNPs and result in a lot of strong oxidative stress.35 Smaller sized AgNPs can produce greater amounts of hydrogen peroxide and induce greater inflammasome formation as they can cause stronger leakage of cathepsins from lysosomes, encourage extra efflux of intracellular K+ and produce superoxide in the membranes of mitochondria.36
2.2.3. Nitric oxide scavenging activity. NO is an important bioregulatory molecule required for many biological processes like the immune response, neural signal transmission, control of blood pressure and regulation of vasodilation.37,38 The biosynthesized AgNPs showed a concentration dependent activity in the NO scavenging assay and the best scavenging activity of 80.46% was observed at a higher concentration of 100 μg mL−1 (Fig. 5c). The above observed NO activity was less than that of the standard BHT (89.35%). It may be that NO was scavenged in extra neuronal tissues and the presence of scavenged NO has been histochemically demonstrated in the adjacent tissues.39
2.2.4. The reducing power. For the biosynthesized AgNPs of the stem/bark extracts, their reducing power increased correspondingly with increase of the concentration of the AgNPs (Fig. 5d). With concentrations of 10 μg mL−1 to 100 μg mL−1, the reducing rate increased from 10.98% to 79.12%. From the results, AgNPs exhibited almost equal reducing power capability when compared to standard BHT (87.35%) due to the presence of the steroid sapogenin in the extract. The steroid sapogenin is known to bind metal ions,40 and the binding of metal ions, such as iron, in vivo is an antioxidant action in itself, preventing the metal ion catalyzed initiation of reactive species.41 However, the steroid sapogenin also has an electron-donating antioxidant capacity.42 This result correlated with biosynthesized AgNPs of Cleistanthus collinus.15
2.3. Antimicrobial activity
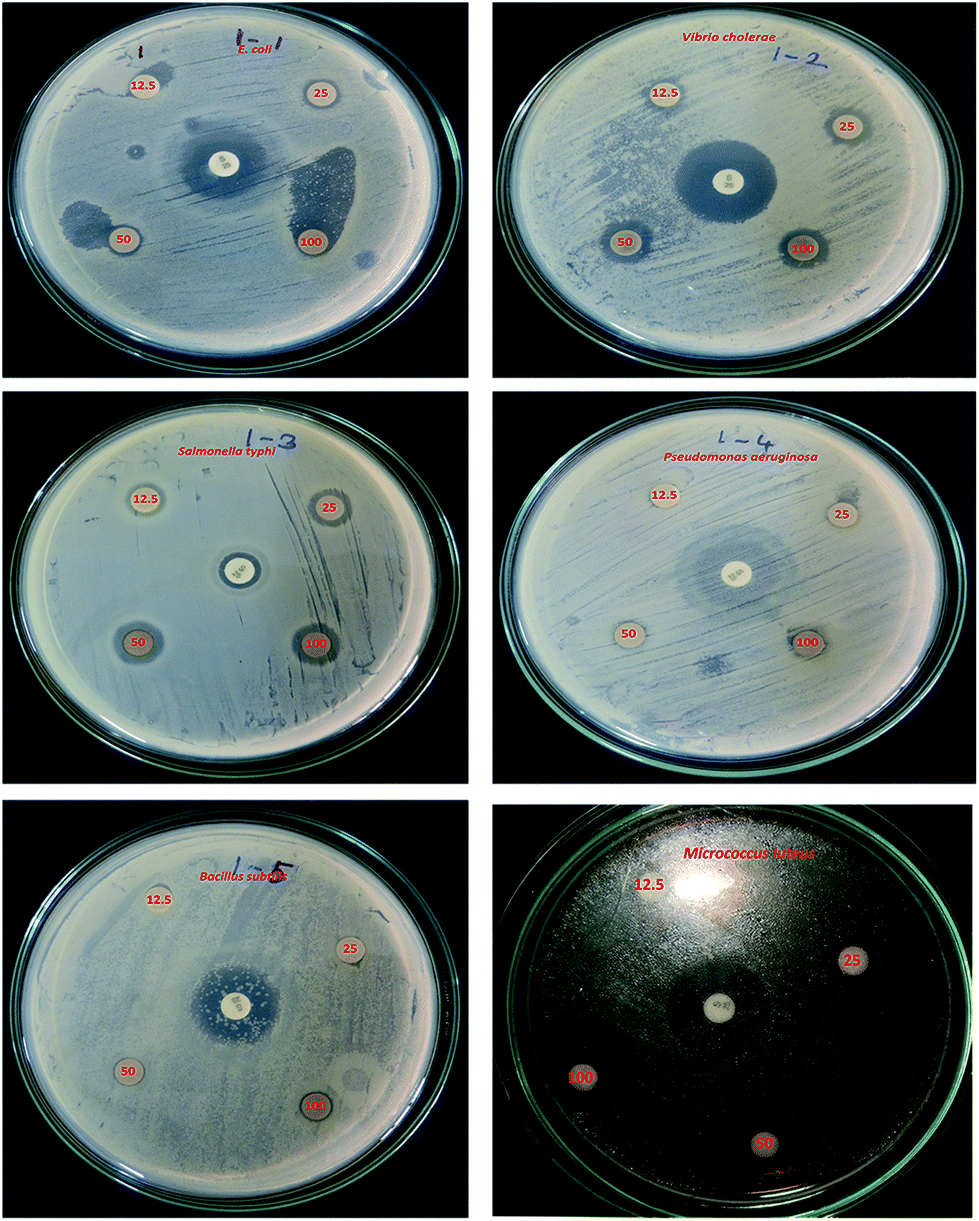
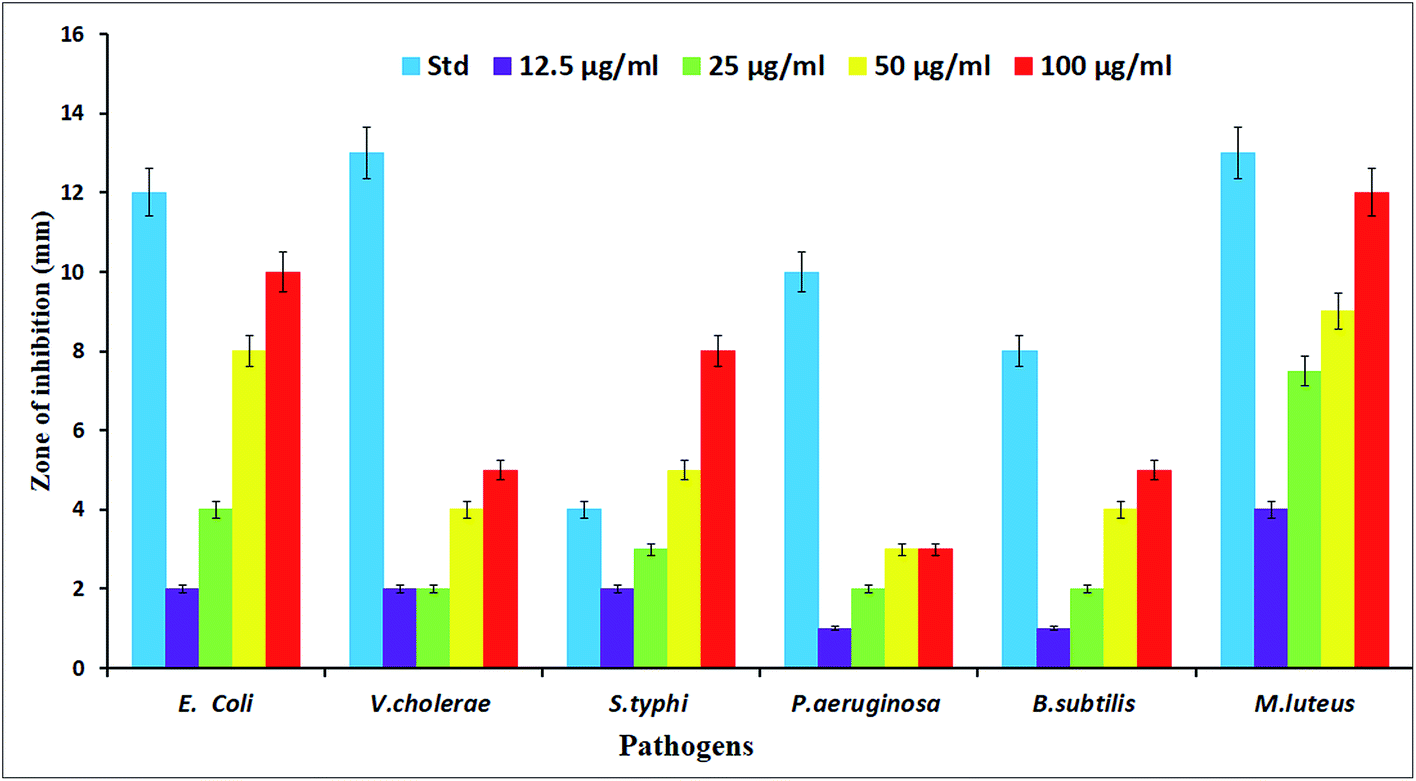
The potential antibacterial activity of the AgNPs was tested against six different pathogenic strains, E. coli, V. cholera, S. typhi, P. aeruginosa, B. subtilis and M. luteus, and compared to a standard antibiotic streptomycin (Fig. 6). The synthesized AgNPs were found to show good antibacterial activity against all six bacterial strains in a dose dependent manner. At a higher (100 μg mL−1) concentration, the AgNPs exhibited a well defined inhibitory effect and produced clearance zones of 10 mm, 5 mm, 8 mm, 3 mm, 5 mm and 12 mm respectively against E. coli, V. cholera, S. typhi, P. aeruginosa, B. subtilis and M. luteus, where the activity was equal to that of streptomycin, which produced clearance zones of 12 mm, 13 mm, 4 mm, 10 mm, 8 mm and 13 mm respectively (Fig. 7). On the other hand, with S. typhi, the AgNPs and streptomycin exhibited 8 mm and 4 mm zones of inhibition respectively at a concentration of 100 μg mL−1. A possible mechanism of the antibacterial activity involves the release of silver cations from the silver nanoparticles that act as reservoirs for the Ag+ bactericidal agent. The interaction of silver cations with bacterial cell membranes, thereby resulting in disturbance of the membrane permeability and of the respiratory system, leads to cell death.43,44
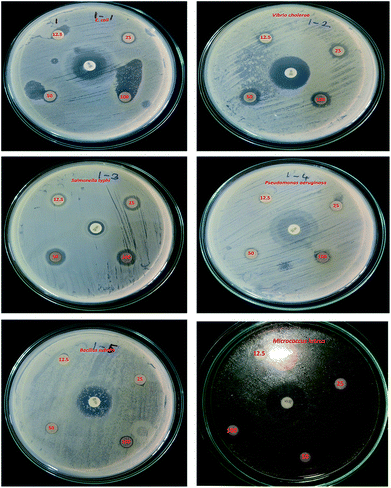 |
| | Fig. 6 Bactericidal effect against six pathogens, using concentrations of 12.5 μg mL−1, 25 μg mL−1, 50 μg mL−1 and 100 μg mL−1. The standard antibiotic is in the centre. | |
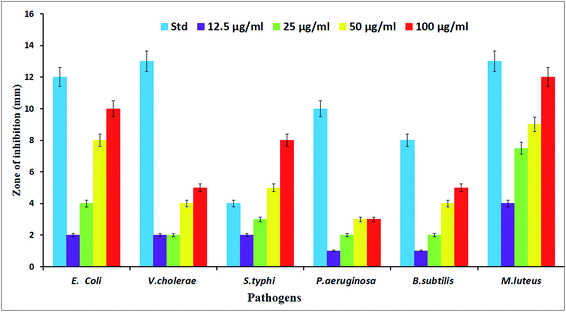 |
| | Fig. 7 Antimicrobial activity of the synthesized AgNPs against various pathogenic bacterial strains. | |
2.4. In vitro anticancer studies
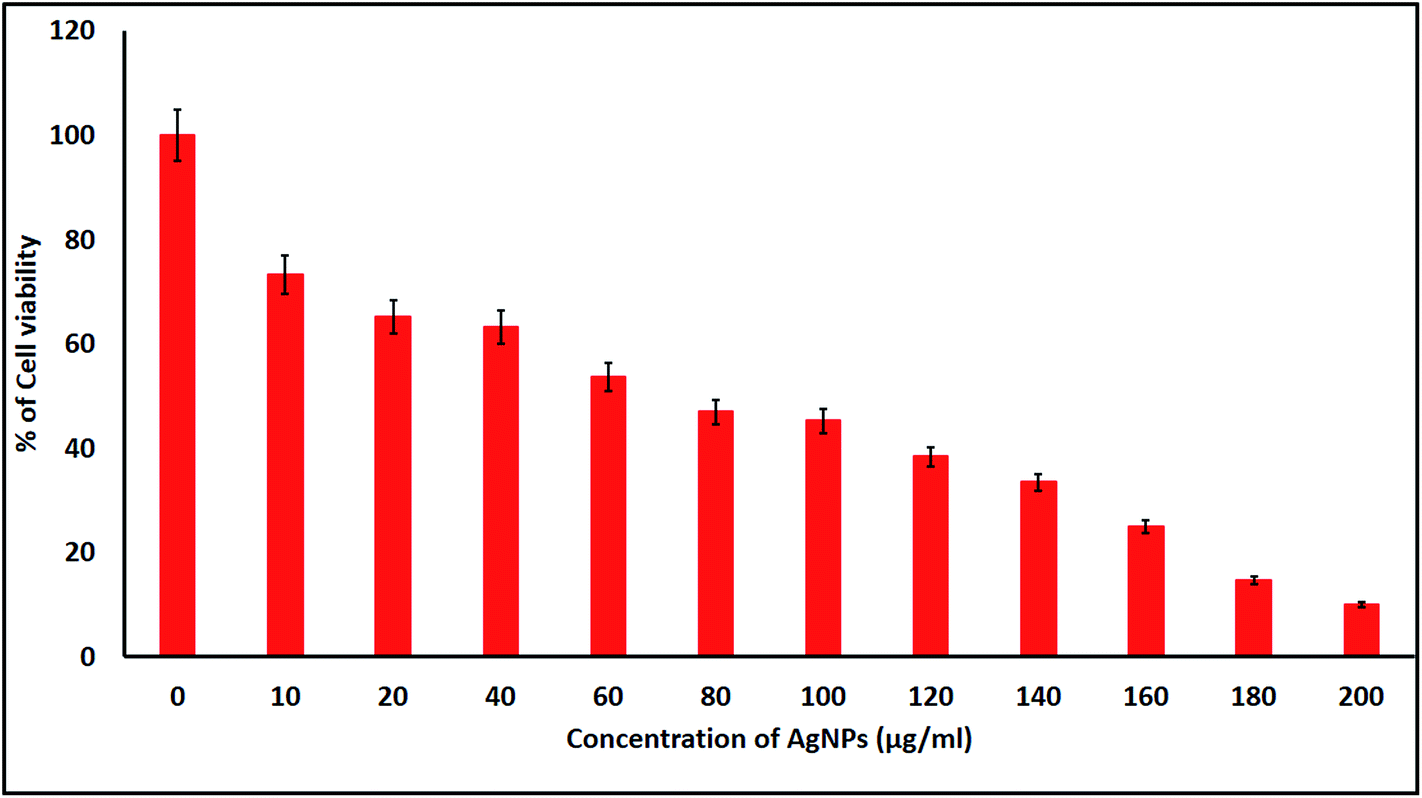
2.4.1. Cell viability and cytomorphological changes of KB cells. KB cells were treated with the biologically synthesized AgNPs at different concentrations for 24 h and the cell viability was measured using a MTT colorimetric assay. The biogenic AgNP induced cytotoxicity for the KB cell lines was found to be higher with increased concentrations of the AgNPs. In the present study, we found that the induction of apoptosis and dose dependent cytotoxicity were observed in the AgNP treated KB cells. The IC50 of the biosynthesized AgNPs was found to be 70 μg mL−1 against the KB cells after 24 h of exposure (Fig. 8). Similar results in terms of the cytotoxicity rates have been discussed by Renugadevi and Aswini.45 In the AgNP treated KB cells, morphological variations were also observed, changes that were not seen in the untreated cells. Morphological differences such as inhibition of cell progress, loss of membrane integrity, cytoplasmic reduction and cell clumping were observed among the NP treated cells. These results show that AgNP treated KB cells undergo cell death, whereas non-treated cells remain viable, at the tested concentrations.
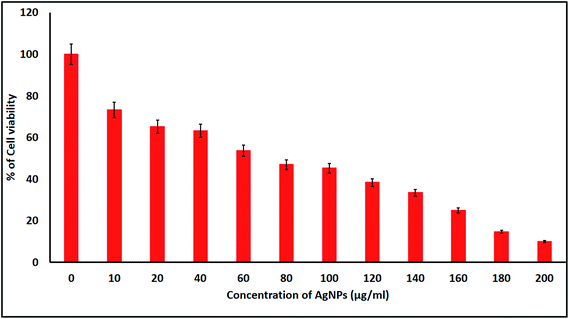 |
| | Fig. 8 MTT assay results confirming the in vitro cytotoxic effect of the AgNPs against KB cells after 24 h. The detected IC50 concentration was 70 μg mL−1 for KB cells after 24 h. Data is expressed as mean ± SD of three experiments. | |
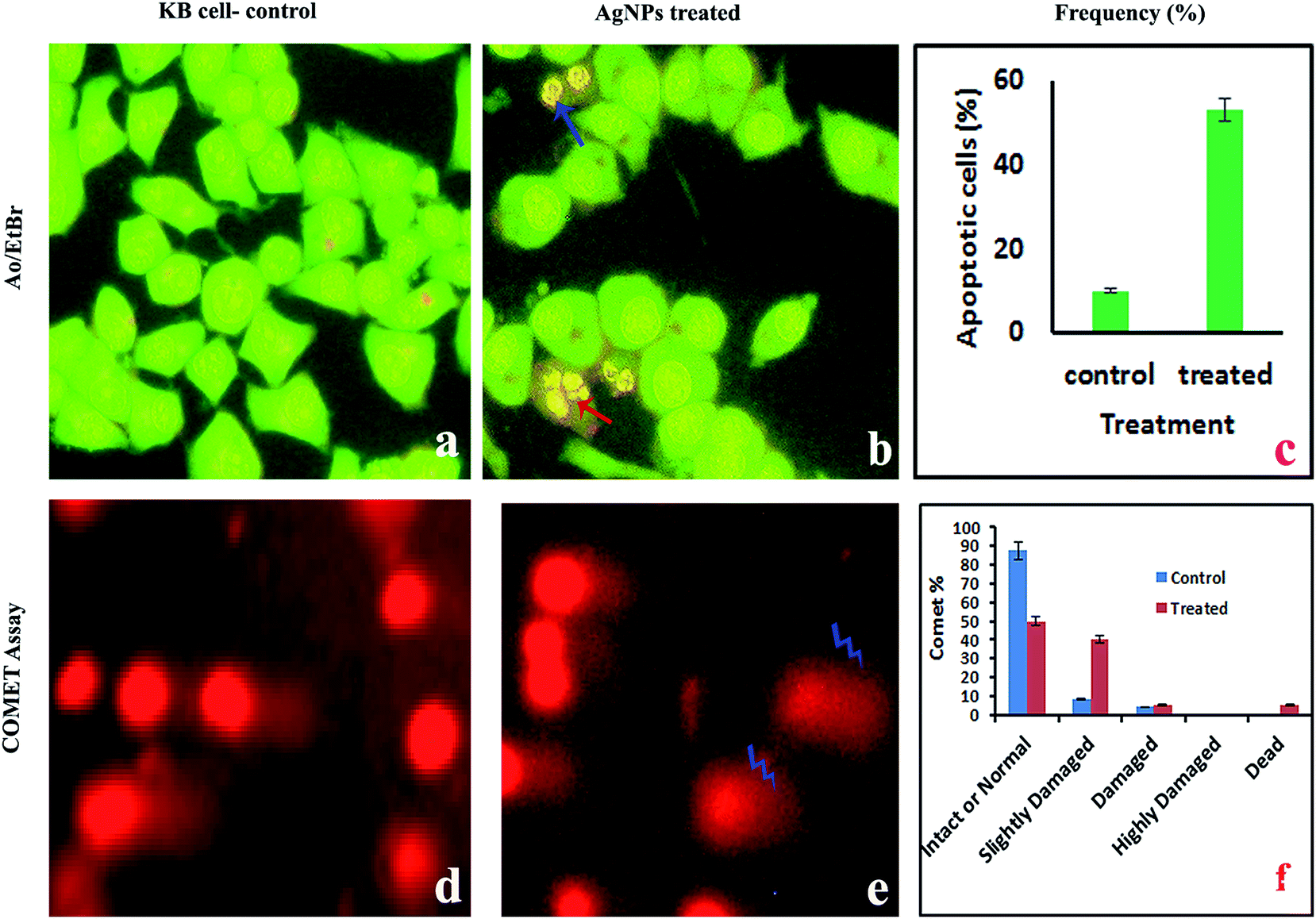
2.4.2. AO/EtBr staining for the detection of apoptotic cells. The pro-apoptotic properties of the AgNPs during treatment of KB cells with the IC50 concentration were studied using fluorescence microscopy after AO/EtBr staining. Since AO can permeate normal cell membranes, nuclei are observed as a green color. EtBr is an intercalating agent that is engaged by cells when their cytoplasmic membrane integrity is lost, and the nuclei are visualized as red in color under fluorescence microscopy. Staining with AO and EB showed significant fragmentation and condensation of the chromatin in KB cells treated with the AgNPs for 24 h (Fig. 9a–c). While the control cells exhibited a normal green nucleus characteristic of diffused chromatin, early apoptotic cells have condensed/fragmented chromatin with a bright green nucleus. Early apoptotic cells show nuclei in an orange color and condensed and fragmented chromatin when viewed under a fluorescence microscope, according to Vivek et al.46
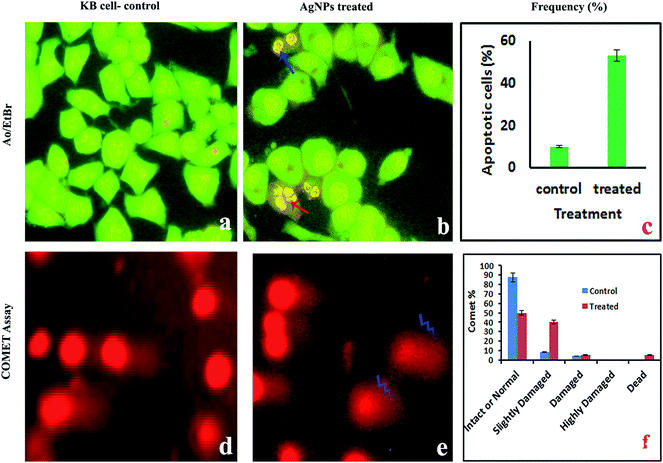 |
| | Fig. 9 Fluorescence microscopy images of KB cells treated with the IC50 concentration of AgNPs. Fluorescence microscopy study with AO/EtBr staining: (a) control cells appear as live cells in a green color; (b) orange colored apoptotic cells indicated by arrows; (c) percentage of apoptosis. Comet assay: (d and e) photomicrographs of DNA damage and (f) the percentage of intact or normal, slightly damaged, damaged, highly damaged and dead cells. | |
2.4.3. Alkaline single-cell gel electrophoresis (comet assay). The AgNP pre-treated cells showed DNA damage, such as fragmented DNA tails, olive tails, altered tail lengths and DNA movement in the KB cells (Fig. 9d and e). In the comet assay during single cell gel electrophoresis, the control cells exhibited mostly reduced fragmentation and unbroken nuclei, whereas tail DNA was observed in the treated cells. The comet is a suitable assay for assessing tumor cell DNA damage and the induction of DNA single strand breaks. Fig. 8f shows the percentage of intact or normal, slightly damaged, damaged, highly damaged and dead cells for the treated and normal KB cells.
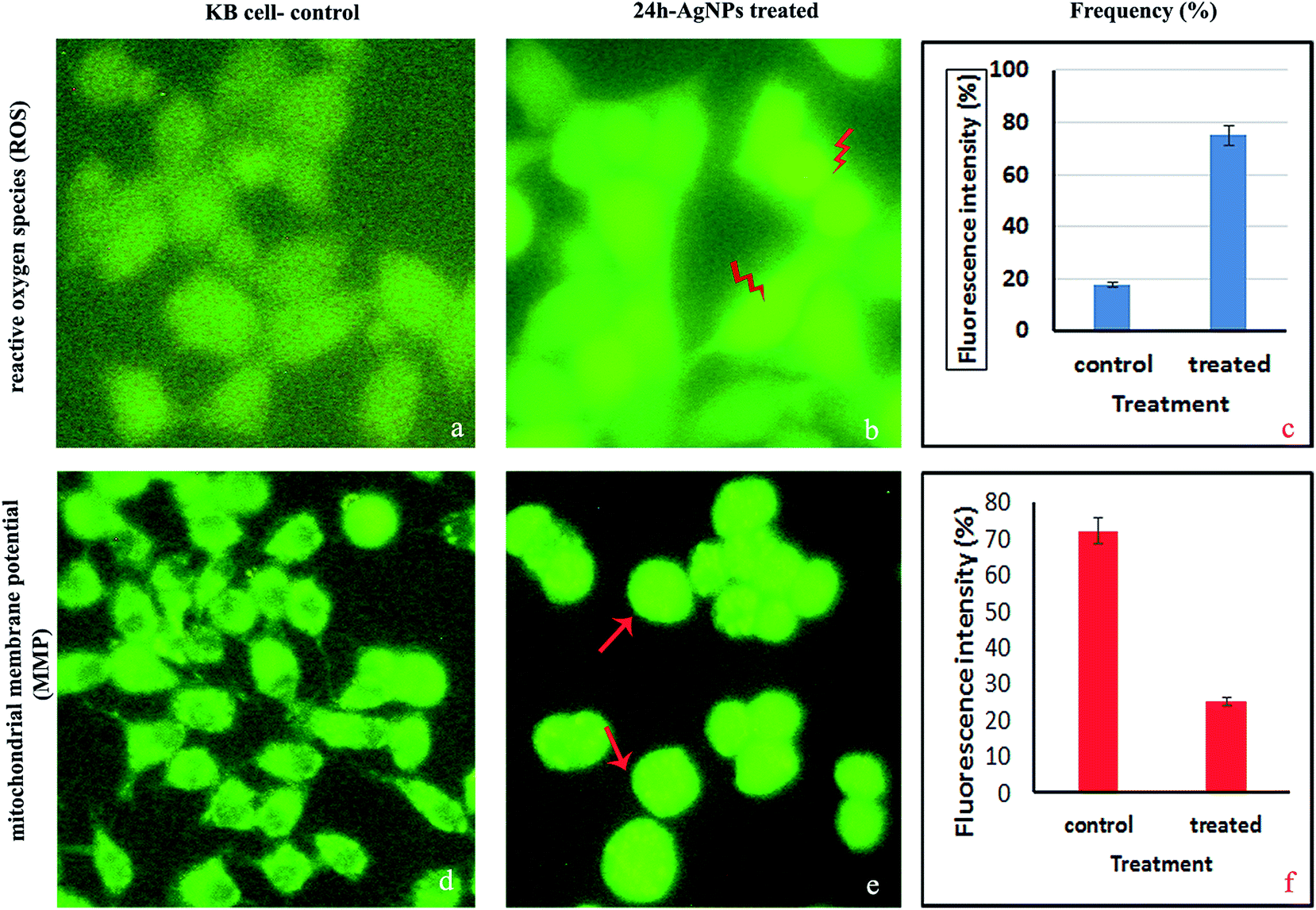
2.4.4. Measurement of the intracellular reactive oxygen species (ROS). The DCFH-DA staining procedure is an indirect method for assessing ROS by measuring intracellular hydrogen peroxide generation. In the present study, the amount of ROS, determined through the fluorescence intensity, was higher for the KB cells treated with AgNPs at the IC50 concentration than in the control non-treated KB cells (Fig. 10a–c). Due to the accumulation of AgNPs in live cell mitochondria, the intracellular ROS concentration was higher for the treated KB cells compared to the untreated cells. This increased level of ROS may be due to cells undergoing the activation of early apoptosis by oxidative stress. ROS are free radicals that play crucial functions in organisms, and in a biological system they help regulate the activity of certain enzymes involved in the apoptotic pathways. Excessive amounts of intracellular reactive oxygen species can cause oxidative damage to proteins, lipids and DNA, but a moderate level of ROS can stimulate cell differentiation and proliferation. Therefore, maintaining a cellular ROS homoeostasis is essential for normal cell growth and survival.47 Studies show that the internal or external addition/generation of ROS can cause cell death by necrosis or apoptosis.48 In our study, the significant production of ROS on AgNP exposure may be assumed to be related to the contribution of oxidative attacks to KB cell death.49
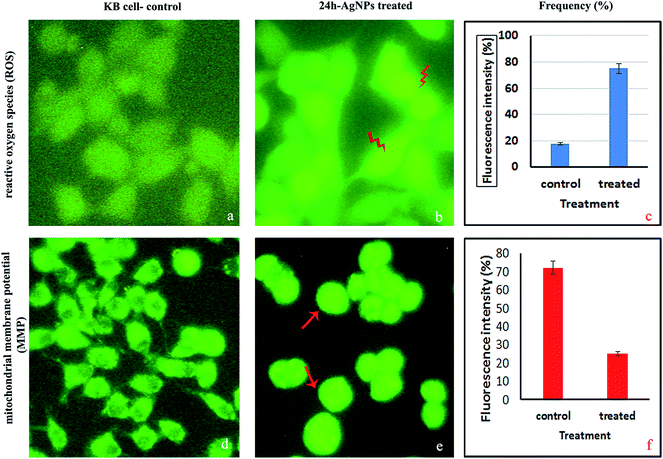 |
| | Fig. 10 Fluorescence microscopy images of KB cells treated with the IC50 concentration of AgNPs. Intracellular reactive oxygen species (ROS): (a) DCFH-DA staining; (b) high DCF fluorescence after AgNP treatment; (c) measurement of the intracellular ROS. Mitochondrial membrane potential (MMP): (d) Rh-123 staining; (e and f) MMP alteration using a spectrofluorimeter. | |
2.4.5. Analysis of the mitochondrial membrane potential (MMP). The MMP was measured using Rh-123 fluorescence and KB cells treated with an IC50 concentration of AgNPs for 24 h (Fig. 10d–f). Live cell mitochondria are capable of retaining this fluorescent dye due to polarization, while dead or apoptotic cells lose this capability because of loss of the mitochondrial membrane integrity and thus, exhibit an exudation of the dye.50 Because mitochondria are known to be involved in the process of programmed cell death, we investigated changes in the MMP in terms of the Rh-123 fluorescence intensity. Fluorescence microscopy images confirmed depletion of the MMP and increased fluorescence intensity after Rh-123 immersion, which reflects depolarization in the AgNP treated cells. MMP loss and an increased level of ROS might be the mechanisms involved in apoptosis induction.48 In this study, the potential role of oxidative stress as a mechanism of AgNP induced apoptosis via the mitochondrial pathway was also studied. Collectively, our findings support the in vitro anticancer activity of the biosynthesized AgNPs and their potential utilization as colloidal drugs and anticancer agents.
2.5. Cytotoxicity studies
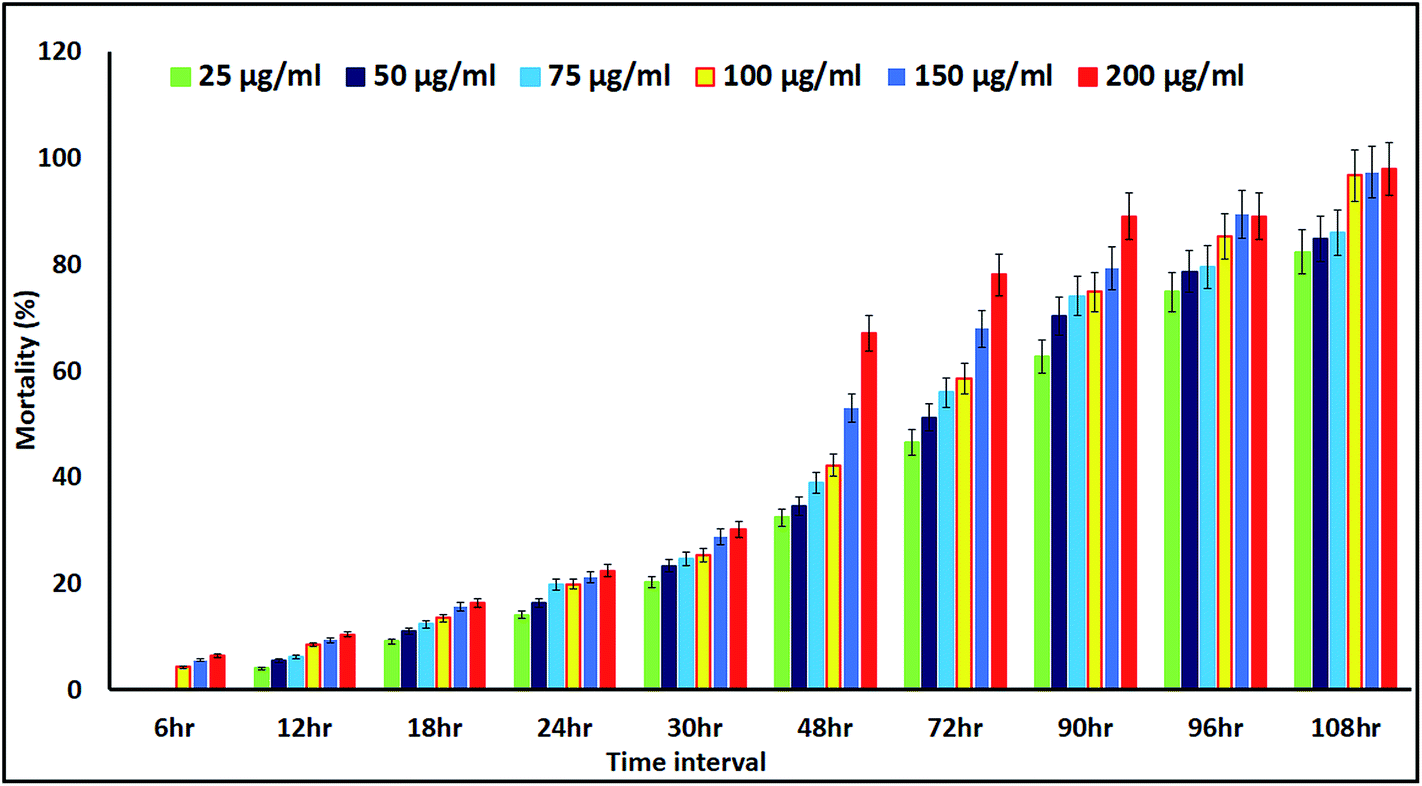
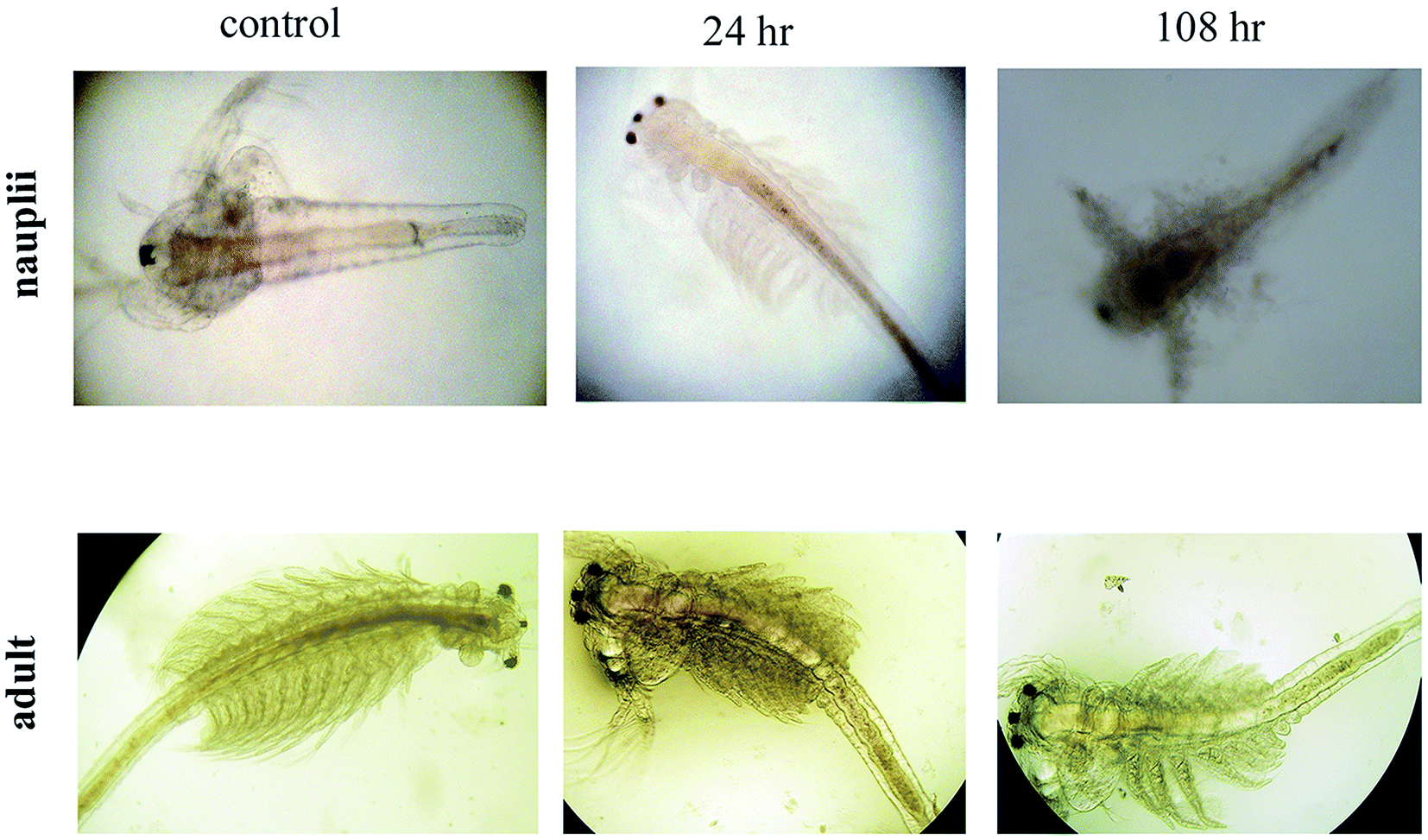
The mortality values are shown in Fig. 11. At 6 h, NP treatment resulted in a 4.23–6.47% mortality rate, with the controls not affected. The mortality rate was dose and exposure time dependent, for a concentration range of 25 μg mL−1 to 200 μg mL−1 and up to 108 h of exposure, with nauplii and adults. At 108 h, the mortality rates of the nauplii and adults were higher for all the concentrations (Fig. 12a–f). The adult mortality rate was less than for the nauplii (data not shown). The mortality rate for the nauplii rose from zero at 25 μg mL−1 to 6.47% at 200 μg mL−1 after 6 h of exposure. These results show that binary compound suspensions of the AgNPs were not acutely toxic to nauplii at high concentrations (75 μg mL−1), after 72 h of exposure. Ates et al.51 reported NPs were not toxic to nauplii at the optimum concentration and with long exposure times.
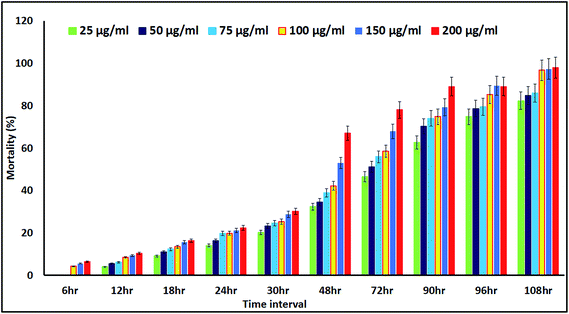 |
| | Fig. 11 Percentage mortality rates for Artemia nauplii, measured for up to 108 h of exposure to different concentrations of the colloidal biosynthesized AgNPs. | |
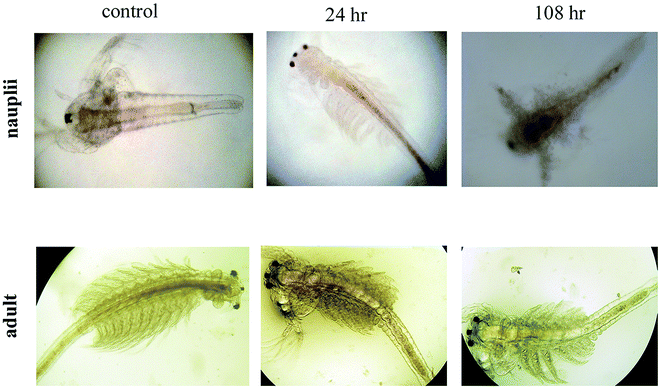 |
| | Fig. 12 Alteration and accumulation of AgNPs in the Artemia adult and nauplii after 24 h and 108 h of exposure. | |
3. Materials and methods
3.1. Plant identification and sample preparation
The H. isora plant was collected from the Western Ghats of Tamil Nadu, India and the species were identified and authenticated by Dr J. H. Benjamin Franklin, Scientist-B. Voucher specimens were prepared and deposited at the herbarium of the Botanical Survey of India (BSI), Southern Regional Centre, Coimbatore. After identification, the stem bark material was collected from the collection site, washed with sterile Milli Q water and dried, and then made into a powder. The plant extracts were prepared according to the Bhakya et al.7 method and stored at 4 °C in a refrigerator until further use.
3.2. Synthesis of the silver nanoparticles (AgNPs)
Silver nanoparticles were synthesized based on the procedure of Bhakya et al.7 The plant extracts were mixed with a 1 mM aqueous AgNO3 solution in a ratio of 1:1 and the mixture was incubated at room temperature for 6 h. Following incubation, the mixture was centrifuged at 18000 rpm for 20 min, and after centrifugation the product was washed with Milli Q water and collected into a collecting chamber for air drying.
3.3. Characterization of the AgNPs
3.3.1. UV-visible spectral analysis. The biosynthesized AgNPs were analyzed using a UV-visible spectrophotometer (UV-1700 Shimadzu, Japan) in the 200–800 nm range at regular 1 h intervals for up to 6 h.
3.3.2. Particle size analysis. The biosynthesized colloidal AgNPs were analyzed using a DLS particle size analyzer [ZETA Seizers Nanoseries (Malvern Instruments Nano ZS)] to determine the size distribution of the nanoparticles.
3.3.3. FTIR spectral analysis. Dry powders of the stem bark extracts and AgNPs were subjected to FTIR spectrometry (Thermo Nicolet Nexus 670 equipped with KBr optics and a DTGS detector) to identify the biomolecules in the stem bark extracts and those associated with the AgNPs. The equipment was operated with a resolution of 4 cm−1 and a scanning range of 500 to 4000 cm−1.
3.3.4. X-ray diffraction (XRD) study. The stem bark extracts embedded with AgNPs were subjected to lyophilization and were powdered for XRD analysis. The diffraction intensities were recorded between 10 and 80° at 2θ angles. The size of the AgNPs was calculated using the following Debye–Scherrer equation
K – shape factor; λ – X-ray wavelength; β – half the maximum intensity (FWHM) in radians; θ – Bragg’s angle.
3.3.5. EDX analysis. In this experiment, a thin film of dry powder of the biosynthesized AgNPs was prepared on a carbon grid (copper coated), dried under a mercury lamp for 5–10 min and analyzed using an EDX analyzer (Hitachi S-4500) to obtain the elemental composition.
3.3.6. Transmission electron microscopy (TEM) study. To determine the size and morphology of the AgNPs, 50 μL of the colloidal solution was diluted to 0.5 mL with Millipore Milli-Q water, sonicated using an ultrasonic bath and filtered with a 0.22 μm filter (Rankem Faridabad, India), and then a drop was placed on a Cu grid and dried under vacuum. Then the NPs were visualized using a JEOL JEM2100 TEM at an acceleration voltage of 200 kV.
3.4. Antibacterial assay
A disc diffusion method was used to determine the antibacterial activity of the biosynthesized AgNPs using the following reference strains: E. coli (ATCC25922), Pseudomonas aeruginosa (ATCC27853), Salmonella typhi (ATCC2426), Vibrio cholera (ATCC2077), Bacillus subtilis (ATCC25923) and Micrococcus luteus (ATCC2077). The overnight inoculated bacterial cultures were spread over freshly prepared Muller-Hinton agar plates. Sterile discs (6 mm, Himedia) containing different concentrations of the silver nanoparticles (12.5, 25, 50 and 100 μg mL−1) were applied and a reference disc of streptomycin was also kept on the plate and incubated at 37 °C for 24 h. The antimicrobial activity of the AgNPs was estimated by measuring the diameter (in millimeters) of the zone of inhibition around the discs after incubation.
3.5. In vitro antioxidant assays
The antioxidant activity of the AgNPs (10–100 μg mL−1) was determined using a DPPH assay according to the method introduced by Brand-Williams et al.52 The DPPH free radical scavenging activity of the biosynthesized AgNPs is expressed as % inhibition of DPPH and was calculated using the following formula:
where Ac is the control absorbance of the DPPH radicals + methanol; As is the absorbance of the sample/the standard BHT.
The hydrogen peroxide (H2O2) radical scavenging activity of the synthesized AgNPs (10–100 μg mL−1) was quantified using the method devised by Pick and Mizel.53 The total reducing power of the synthesized AgNPs (10–100 μg mL−1) was estimated by following the method devised by Oyaizu.54 The nitric oxide scavenging activity of the synthesized AgNPs (10–100 μg mL−1) was estimated using the Sousa et al.55 reaction method. The obtained results were calculated using the above mentioned formula and compared with the standard antioxidant butylated hydroxytoluene (BHT).
3.6. In vitro anticancer studies
3.6.1. Cytotoxicity assay. The cytotoxicity of the biosynthesized AgNPs against an oral carcinoma (KB) line was measured using a MTT calorimetric assay (TC191, Himedia, Mumbai, India). KB cell lines were sown in 96 well plates at a density of 10000 cells per well and kept in an incubator for up to 24 h for acclimatization. After 24 h, the cells were treated with various concentrations (10, 20, 40, 60, 80, 100, 120, 140, 160, 180 and 200 μg mL−1) of the AgNPs, along with a control, following a procedure by Dash et al.56 For the cell viability study, the absorbance was measured at 540 nm using a microplate reader. The required concentrations for 50% inhibition of the viability (IC50) were calculated graphically and the optimum dose was used for further studies. Determination of the cell morphology was performed using a phase contrast microscope (Nikon, Eclipse TS100, Japan).
3.6.2. Acridine orange (AO) and ethidium bromide (EtBr). Apoptotic morphological changes were investigated using a AO/EtBr staining procedure, according to a report by Vivek et al.46
3.6.3. Alkaline single-cell gel electrophoresis (comet assay). Seeded cells in 96-well plates were treated with or without AgNPs at 70 μg mL−1 for 48 h. Cells were then harvested and the DNA damage was determined with a comet assay. This assay was carried out to estimate the DNA damage caused by the AgNPs based on the protocol of Singh et al.57
3.6.4. Measurement of the intracellular reactive oxygen species (ROS). Based on the oxidative conversion of 2′,7′-dichlorofluorescein-diacetate (DCFH-DA) to the fluorescent compound dichloroflouorescin (DCF), intracellular ROS formation was measured based on the method by Roy et al.58
3.6.5. Analysis of mitochondrial membrane potential (MMP). The early stages of apoptosis are indicated by an alteration in the mitochondrial membrane potential. Rhodamine 123 (Rh-123) is a cationic, green-fluorescent dye, and it was used to detect changes in the mitochondrial membrane potential on exposure to the AgNPS.56 Treated cells were viewed under a fluorescence microscope using a blue filter.
3.7. Brine shrimp toxicity study
Brine shrimp (Artemia salina) were hatched using the procedure described by Persoone et al.59 After hatching, counting was performed according to the procedure described by Sorgeloos.60 A study of the cytotoxicity of the AgNPs was conducted using nauplii for 6 h to 108 h. For the experiment, 100 nauplii were placed in 6 well plates, each well containing 20 mL of sea water and a different concentration of the AgNPs (25, 50, 75, 100, 150 and 200 μg mL−1), and were maintained at room temperature for 108 h under light conditions. The mortality rates were then calculated. The experiments were conducted along with a control, and every experiment was carried out in triplicate.
4. Conclusion
A stable, simple and eco-friendly technique for biosynthesizing AgNPs was effectively established using H. isora stem bark extract. H. isora stem bark contains more phytosterols and saponins, which play major roles as reducing as well as capping agents in the green synthesis of AgNPs. The extract acts as a reducing and stabilizing agent, which was confirmed through FTIR studies. TEM and XRD results revealed that the biosynthesized AgNPs were crystalline in nature with a mean particle size of 25.55 nm. The biosynthesized AgNPs were found to be multifunctional with good antioxidant and antimicrobial activities. The stem bark extract capped AgNP solution showed a significant dose-dependent cytotoxic effect on KB cells. Nauplii showed significant mortality after 108 h of exposure to the AgNPs. Thus, it was concluded that the colloidal AgNPs are not acutely toxic to Artemia. This green synthesis method might be a good alternative to physical and chemical nanosynthesis methods. Hence, this method might be scaled up for industrial production and the manufacturing of ultrafiltration membranes as well as food packaging, and might be of value for many biological and medical applications.
Acknowledgements
The work was financially supported by a UGC-Rajiv Gandhi National Fellowship (RGNF) No: F1-17.1/2013-14/RGNF-2013-14-SC-TAM-44942 (SA-III) University Grant Commission New Delhi, India of the first author. We thank the sophisticated analytical instrument facility (SAIF), North-Eastern Hill University (NEHU), Shillong for access to the TEM facility. The authors wish to thank the following individuals who provided valuable advice in the final stage of the revision process: Dr V. Ramalingam, Department of Marine Science, Bharathidasan University, and M. Muthukumar and S. Parthibhan, Research Scholars (Department of Plant Science, Bharathidasan University, Tiruchirappalli-620 024, Tamil Nadu, India), and S. Arul Ganesh, Dr S. Karthikeyeni and S. Arun (Research Scholars, Department of Animal Science, Bharathidasan University, Tiruchirappalli-620 024, India).
References
- Z. U. Khan, WebmedCentral CANCER, 2012, 3, WMC003626 Search PubMed.
- R. L. Kruse, J. R. Kramer, G. L. Tyson, Z. Duan, L. Chen, H. B. El-Serag and F. Kanwal, Hepatology, 2014, 60, 1871–1878 CrossRef PubMed.
- C. G. Kumar and Y. Poornachandra, Colloids Surf., B, 2015, 125, 110–119 CrossRef CAS PubMed.
- J. Sekuła, J. Nizioł, W. Rode and T. Ruman, Anal. Chim. Acta, 2015, 875, 61–72 CrossRef PubMed.
- T. Gunasekaran, T. Nigusse and M. D. Dhanaraju, J. Am. Coll. Clin. Wound Spec., 2011, 3, 82–96 CrossRef PubMed.
- B. Godin, J. H. Sakamoto, R. E. Serda, A. Grattoni, A. Bouamrani and M. Ferrari, Trends Pharmacol. Sci., 2010, 31, 199–205 CrossRef CAS PubMed.
- S. Bhakya, S. Muthukrishnan, M. Sukumaran and M. Muthukumar, Appl. Nanosci., 2015, 1–12 CAS.
- S. Muthukrishnan, S. Bhakya, T. S. Kumar and M. Rao, Ind. Crops Prod., 2015, 63, 119–124 CrossRef CAS.
- S. Bhakya, S. Muthukrishnan, M. Sukumaran, M. Muthukumar, S. Kumar and M. Rao, J. Biorem. Biodegrad., 2015, 6, 312 Search PubMed.
- T. Sinha, M. Ahmaruzzaman and A. Bhattacharjee, J. Environ. Chem. Eng., 2014, 2, 2269–2279 CrossRef CAS.
- A. C. Joshi, G. B. Markad and S. K. Haram, Electrochim. Acta, 2015, 161, 108–114 CrossRef CAS.
- M. El-Rafie, T. I. Shaheen, A. Mohamed and A. Hebeish, Carbohydr. Polym., 2012, 90, 915–920 CrossRef CAS PubMed.
- M. Jalali, S. Dauterstedt, A. Michaud and R. Wuthrich, Composites, Part B, 2011, 42, 1420–1426 CrossRef.
- E. Wetzel, http://nano--tech.blogspot.in/p/military.html, 2015.
- N. Kanipandian, S. Kannan, R. Ramesh, P. Subramanian and R. Thirumurugan, Mater. Res. Bull., 2014, 49, 494–502 CrossRef CAS.
- A. Melaiye and W. J. Youngs, Expert Opin. Ther. Pat., 2005, 15, 125–130 CrossRef CAS.
- D. G. Maki, Clin. Infect. Dis., 2010, 50, 1580–1587 CrossRef PubMed.
- M. Zargar, K. Shameli, G. R. Najafi and F. Farahani, J. Ind. Eng. Chem., 2014, 20, 4169–4175 CrossRef CAS.
- R. Arunachalam, S. Dhanasingh, B. Kalimuthu, M. Uthirappan, C. Rose and A. B. Mandal, Colloids Surf., B, 2012, 94, 226–230 CrossRef CAS PubMed.
- G. Sathishkumar, C. Gobinath, K. Karpagam, V. Hemamalini, K. Premkumar and S. Sivaramakrishnan, Colloids Surf., B, 2012, 95, 235–240 CrossRef CAS PubMed.
- K. Niraimathi, V. Sudha, R. Lavanya and P. Brindha, Colloids Surf., B, 2013, 102, 288–291 CrossRef CAS PubMed.
- H. Jiang, K.-s. Moon, Z. Zhang, S. Pothukuchi and C. Wong, J. Nanopart. Res., 2006, 8, 117–124 CrossRef CAS.
- A. Ahmad, P. Mukherjee, S. Senapati, D. Mandal, M. I. Khan, R. Kumar and M. Sastry, Colloids Surf., B, 2003, 28, 313–318 CrossRef CAS.
- V. Ramalingam, R. Rajaram, C. PremKumar, P. Santhanam, P. Dhinesh, S. Vinothkumar and K. Kaleshkumar, J. Basic Microbiol., 2014, 54, 928–936 CrossRef CAS PubMed.
- M. Kowshik, N. Deshmukh, W. Vogel, J. Urban, S. K. Kulkarni and K. Paknikar, Biotechnol. Bioeng., 2002, 78, 583–588 CrossRef CAS PubMed.
- R. Mata, J. R. Nakkala and S. R. Sadras, Colloids Surf., B, 2015, 128, 276–286 CrossRef CAS PubMed.
- C. Krishnaraj, E. Jagan, S. Rajasekar, P. Selvakumar, P. Kalaichelvan and N. Mohan, Colloids Surf., B, 2010, 76, 50–56 CrossRef CAS PubMed.
- D. E. Sands, Introduction to crystallography, Courier Corporation, 2012 Search PubMed.
- T. J. I. Edison and M. Sethuraman, Process Biochem., 2012, 47, 1351–1357 CrossRef CAS.
- T. Prathna, N. Chandrasekaran and A. Mukherjee, Colloids Surf., A, 2011, 390, 216–224 CrossRef CAS.
- J. Kasthuri, S. Veerapandian and N. Rajendiran, Colloids Surf., B, 2009, 68, 55–60 CrossRef CAS PubMed.
- A. M. Fayaz, K. Balaji, M. Girilal, R. Yadav, P. T. Kalaichelvan and R. Venketesan, Nanomedicine: Nanotechnology, Biology and Medicine, 2010, 6, 103–109 CrossRef CAS PubMed.
- T. Suman, S. R. Rajasree, A. Kanchana and S. B. Elizabeth, Colloids Surf., B, 2013, 106, 74–78 CrossRef CAS PubMed.
- M. R. Mozafari, J. Flanagan, L. Matia-Merino, A. Awati, A. Omri, Z. E. Suntres and H. Singh, J. Sci. Food Agric., 2006, 86, 2038–2045 CrossRef CAS.
- W. He, Y.-T. Zhou, W. G. Wamer, M. D. Boudreau and J.-J. Yin, Biomaterials, 2012, 33, 7547–7555 CrossRef CAS PubMed.
- E.-J. Yang, S. Kim, J. S. Kim and I.-H. Choi, Biomaterials, 2012, 33, 6858–6867 CrossRef CAS PubMed.
- A. Gibson, Eur. J. Pharmacol., 2001, 411, 1–10 CrossRef CAS PubMed.
- D. Rees, R. Palmer and S. Moncada, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 3375–3378 CrossRef CAS.
- D. S. Bredt, P. M. Hwang and S. H. Snyder, Nature, 1990, 347, 768–770 CrossRef CAS PubMed.
- N. J. Miller, C. Castelluccio, L. Tijburg and C. Rice-Evans, FEBS Lett., 1996, 392, 40–44 CrossRef CAS PubMed.
- I. Emerit, Free Radicals Biol. Med., 1994, 16, 99–109 CrossRef CAS PubMed.
- Y.-L. Lin, I.-M. Juan, Y.-L. Chen, Y.-C. Liang and J.-K. Lin, J. Agric. Food Chem., 1996, 44, 1387–1394 CrossRef CAS.
- X.-x. Dong, Y. Wang and Z.-h. Qin, Acta Pharmacol. Sin., 2009, 30, 379–387 CrossRef CAS PubMed.
- E. Paszek, J. Czyz, O. Woźnicka, D. Jakubiak, J. Wojnarowicz, W. Łojkowski and E. Stępień, J. Biomed. Nanotechnol., 2012, 8, 957–967 CrossRef CAS PubMed.
- K. Renugadevi and R. V. Aswini, Int. J. Nanomater. Biostruct., 2012, 2, 5–10 Search PubMed.
- R. Vivek, R. Thangam, K. Muthuchelian, P. Gunasekaran, K. Kaveri and S. Kannan, Process Biochem., 2012, 47, 2405–2410 CrossRef CAS.
- P. D. Ray, B.-W. Huang and Y. Tsuji, Cell. Signalling, 2012, 24, 981–990 CrossRef CAS PubMed.
- M. Jeyaraj, M. Rajesh, R. Arun, D. MubarakAli, G. Sathishkumar, G. Sivanandhan, G. K. Dev, M. Manickavasagam, K. Premkumar and N. Thajuddin, Colloids Surf., B, 2013, 102, 708–717 CrossRef CAS PubMed.
- P. M. G. Nair, S. Y. Park and J. Choi, Chemosphere, 2013, 92, 592–599 CrossRef CAS PubMed.
- A. Jones, J. Van Blerkom, P. Davis and A. A. Toledo, Hum. Reprod., 2004, 19, 1861–1866 CrossRef PubMed.
- M. Ates, J. Daniels, Z. Arslan and I. O. Farah, Environ. Monit. Assess., 2013, 185, 3339–3348 CrossRef CAS PubMed.
- W. Brand-Williams, M.-E. Cuvelier and C. Berset, LWT--Food Sci. Technol., 1995, 28, 25–30 CrossRef CAS.
- E. Pick and D. Mizel, J. Immunol. Methods, 1981, 46, 211–226 CrossRef CAS PubMed.
- M. Oyaizu, Jpn. J. Nutr., 1986, 44, 307–315 CrossRef CAS.
- A. Sousa, I. C. Ferreira, L. Barros, A. Bento and J. A. Pereira, LWT--Food Sci. Technol., 2008, 41, 739–745 CrossRef CAS.
- S. K. Dash, S. Chattopadhyay, T. Ghosh, S. Tripathy, S. Das, D. Das and S. Roy, ISRN Oncol., 2013, 2013, 709269 Search PubMed.
- N. P. Singh, M. T. McCoy, R. R. Tice and E. L. Schneider, Exp. Cell Res., 1988, 175, 184–191 CrossRef CAS PubMed.
- A. Roy, A. Ganguly, S. BoseDasgupta, B. B. Das, C. Pal, P. Jaisankar and H. K. Majumder, Mol. Pharmacol., 2008, 74, 1292–1307 CrossRef CAS PubMed.
- G. Persoone, A. Van de Vel, M. Van Steertegem and B. De Nayer, Aquat. Toxicol., 1989, 14, 149–167 CrossRef CAS.
- P. Sorgeloos, Mar. Ecol.: Prog. Ser., 1980, 3, 363–364 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra17569d |
| ‡ Two authors equally contributed. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.