DOI:
10.1039/C6RA17424H
(Paper)
RSC Adv., 2016,
6, 109434-109441
Influence of structure–property relationships of two structural isomers of thiophene-flanked diazaisoindigo on carrier-transport properties†
Received
7th July 2016
, Accepted 31st October 2016
First published on 8th November 2016
Abstract
Based on the electron-accepting building block of 7,7′-diazaisosindigo (DAII), two structural isomers of thiophene-flanked diazaisoindigo, 6,6′-substituted 6,6′-T-DAII for full conjugation and 5,5′-substituted 5,5′-T-DAII for cross-conjugation, have been designed and synthesized to study the influence of the connecting positions of the flanking thiophene on the optoelectronic properties, crystal structures, and solid-state microstructures, being well associated with the carrier-transport properties. The results indicate that the 5,5′-substitution on the DAII core of 5,5-T-DAII stabilizes both the HOMO and LUMO levels and reduces the oscillation strength of the low-energy absorption band as compared to the 6,6′-substitution. In the crystals, 6,6′-T-DAII, 5,5′-T-DAII, and 6,6′-dibromo-DAII adopt a flat platform with co-facial slipped π–π stacking. The organic field-effect transistors based on 6,6′-T-DAII and 5,5′-T-DAII on a tetratetracontane (TTC) modified substrate exhibit an n-dominant ambipolar performance with the hole and electron mobilities of 10−3 cm2 V−1 s−1. Of particular note is that 5,5′-T-DAII show obviously higher carrier-mobilities compared to 6,6′-T-DAII, suggesting that the cross-conjugation of the 5,5′-connection on the DAII core could be designed to improve the carrier-transport properties for FET applications.
Introduction
Organic semiconductors with the advantages of low cost, large-area printing, mechanical flexibility, and tunable electronic structures are of particular interest for the development of organic field-effect transistors (OFETs).1–6 Donor–acceptor (D–A) alternation of π-conjugated semiconductors is commonly used to tune the optoelectronic properties and tighten the intermolecular packing coming from the electron push–pull effect between the donor and acceptor parts, and thereby improve the OFET performance with mobilities over 10 cm2 V−1 s−1 for holes7–9 and 6 cm2 V−1 s−1 for electrons.10,11 Compared to the hole-transporting donor units, the development of the electron-transporting acceptor counterparts is still lagging. Therefore, one of the main challenges is to design and synthesize new electron accepting units to create both small-molecule and polymer semiconductors. Tailoring the energy of the frontier molecular orbitals (FMOs) using electron-deficient moieties to stabilize the LUMO will eventually lead to conjugated systems favouring n-type carrier-transport.12 The representative n-type units composed of electron withdrawing amide/imide structures widely reported to date are arylene diimide (naphthalenediimide (NDI) and perylenediimide (PDI)),13–15 diketopyrrolopyrrole (DPP),15–17 tyrian purple,18 and isoindigo (IID).19–21
IID is a useful building block for organic electronics due to the electron-deficient character constituting the donor–acceptor (D–A) π-conjugated organic semiconductors that show an excellent performance in organic field effect transistors (FETs) and organic photovoltaics (OPVs) (Fig. 1).22 Based on IID, a practical approach to efficiently create a stronger n-type building block than IID is to afford an electron-negativity on the IID core for realizing a low-lying LOMO level. Moreover, for the chemical modification of IID, the connection modes of the substituents, the 6,6′-substitution for full-conjugation and the 5,5′-substitution for cross-conjugation, remarkably alter its structure–property relation to realize a coplanar and rigid backbone as described in previous reports.23–25 When considering the connection patterns of isoindigo to construct small-molecule and polymer semiconductors, the outer benzene ring sometimes forms torsion angles with the connecting counterparts, providing the twisted π-conjugated structure, thereby, chemical modifications of the outer benzene ring strongly influence their molecular conformation and electronic structures.26–32
 |
| | Fig. 1 (a) Chemical structures of isoindigo and 6,6′- and 5,5′-dibromo-7,7′-diazaisoindigos and (b) 6,6′-T-DAII and 5,5′-T-DAII. | |
In this context, we have designed two structural isomers of the 6,6′- and 5,5′-dibromo-7,7′-diazaisoindigo (DAII) as versatile synthetic precursors for the n-type building blocks, wherein the carbons at the 7,7′-positions on the IID core are replaced with electron-negative nitrogens (Fig. 1). These molecules are expected to couple with various counterparts constituting the D–A alternation. We reported, recently, ambipolar organic transistor performance based on IID derivatives including DAII molecules.33 The introduction of electron-negative nitrogens contributes to lowering the LUMO level. Additionally, the position of the nitrogen substitution on IID proves to be intriguing for the planarization of IID, in which the nitrogen replaced at the 7-position on DAII forms a fully flat molecular geometry estimated from the theoretical calculations.34,35 The basic characters of 7,7′-DAII, which have been rarely examined, show a promising building unit for organic electronics.36 Most recently, 7,7′-DAII-based polymers using the 6,6′-D–A alternation exhibit an excellent ambipolar or p-channel FET performance, wherein the hole mobility is over 7 cm2 V−1 s−1.35 For the further advance of DAII-based materials, as well as the electron-deficient character of the DAII unit, the substitution positions should be important to modulate the molecular geometries and electronic structures, enabling a tailored structural design for the device application.
In order to simply interpret the influence of the basic properties of the DAII-based molecules coming from the connection patterns on the carrier-transport properties, we report the design and synthesis of two model molecules of the thiophene-flanked diazaisoindigos, 6,6′-T-DAII and 5,5′-T-DAII, since the electron-rich thiophene is more popular in designing D–A alternating molecules. Additionally, compared to the conventional 6,6′-connection, exploring the not commonly studied 5,5′-connection is instructive. 6,6′-T-DAII and 5,5′-T-DAII were fully characterized by X-ray structure analyses, optical spectroscopy, and cyclic voltammetry. Thin-film transistors made of 6,6′-T-DAII and 5,5′-T-DAII exhibited an n-dominant ambipolar behavior irrespective of the connection modes (both the hole-mobilities and electron mobilities are around 10−3 cm2 V−1 s−1) on the tetratetracontane self-assembled monolayers under vacuum conditions. Most importantly, 5,5′-T-DAII exhibits a higher carrier-transport property than 6,6′-T-DAII. The obtained results provide a guideline for designing novel 5,5′-substituted DAII-based small-molecule and polymer semiconductors for FET applications.
Results and discussion
Theoretical estimation
We initially carried out DFT calculations at the ωB97XD/6-31G(d,p) level to estimate the impact on the optimized structures by replacing two carbon positions with the corresponding nitrogens on the IID core (Fig. S1†).34 Of particular note is that the 7,7′-replacement renders the IID core with an entirely flat geometry as compared to other replacements, which agrees with the results of a recent report.35 This observation is presumably due to reducing the electrostatic repulsions with the enlarged distance between the electron-negative nitrogen and oxygen. To evaluate the influence of the connection patterns of the thiophene-flanked 6,6′-T-DAII and 5,5′-T-DAII on the optimized and electronic structures, we calculated four sets of molecules including the IID-based molecules of 6,6′-T-IID and 5,5′-T-IID for comparison (Fig. 2). For ease of the calculations, the hexyl chains were replaced with methyl groups. 6,6′-T-DAII have an entirely flat platform without any noticeable dihedral angles, whereas 5,5′-T-DAII adopts a slightly distorted DAII core by 6.7° and the dihedral angle between the DAII core and flanking thiophene is 35.6°. In comparison, both 6,6′-T-IID and 5,5′-T-IID are nonplanar; the flanked-thiophenes are twisted from the DAII core with the dihedral angles of around 35.5°, and IID cores are distorted by around 15.7°. The planarized DAII core relative to the IID core, especially as observed in 6,6′-T-DAII, would be associated with the electron-negative nitrogen on the DAII unit, and 6,6′-positionings of the flanking thiophenes contribute to releasing the steric hindrance to make the coplanar π-backbone. Obviously, the HOMO and LUMO levels of 6,6′-T-DAII and 5,5′-T-DAII are lower than those of the corresponding 6,6′-T-IID and 5,5′-T-IID, respectively, which means that the electron-withdrawing nitrogen can affect both the HOMO and LUMO levels. The HOMO and LUMO levels were estimated to be −7.526 eV and −1.664 eV for 6,6′-T-DAII and −7.804 eV and −1.700 eV for 5,5′-T-DAII, respectively. It is noteworthy that the 5,5′-substitution of 5,5′-T-DAII weakens the π-delocalization along the conjugated backbone at the central lactam part, which leads to a deeper HOMO of 5,5′-T-DAII than that of 6,6′-T-DAII. Meanwhile, LUMO of 5,5′-T-DAII are mainly concentrated on the DAII core. This reveals that the cross-conjugation of the 5,5′-connection is less effective for the expanding-π-conjugation as compared to the 6,6′-connection.
 |
| | Fig. 2 Optimized structures and frontier molecular orbitals of (a) 6,6′-T-DAII, (b) 5,5′-T-DAII, (c) 6,6′-T-IID, (d) 5,5′-T-IID. Orbital energies are in eV. | |
Synthesis
The syntheses of the structural isomers 6,6′-T-DAII and 5,5′-T-DAII are outlined in Scheme 1. Hexyl chains are installed to improve the solubility for easy purification. Compound 1 was prepared following to a previous report.37 For the preparation of 7a, 6-brominated azaindole 1 was alkylated with hexyl bromide to afford 2, which was treated with PBPB to obtain 3,38 and subsequent reduction with zinc afforded the 6-brominated-oxiindole 7a. For the preparation of 7b, the commercially-available azaindole 4 was first N-alkylated with hexyl bromide, then bromination with bromine afforded 6,39 which was subjected to selective reduction with zinc to provide the 6-brominated-oxindole 7b. These brominated-oxiazaindoles 7a and 7b were readily converted to the corresponding isatins 8a and 8b, respectively, as previously reported.40 The condensation of the oxindoles 7a and 7b with the corresponding isatins 8a and 8b in refluxing acetic acid in the presence of a catalytic amount of HCl gave the two structural isomers 9a and 9b, respectively. Notably, 9a and 9b are key building blocks since π-elongated backbone could be readily achieved through various coupling reactions. The structural isomers 6,6′-T-DAII and 5,5′-T-DAII were obtained via the Stille coupling with tributylstannyl-thiophene.41
 |
| | Scheme 1 Synthetic route of 6,6′-T-DAII and 5,5′-T-DAII. | |
The newly synthesized DAII derivatives were fully characterized by 1H-NMR, 13C-NMR, mass spectroscopy, and elemental analysis. Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were performed on both molecules. The TGA profiles revealed that the two structural isomers 6,6′-T-DAII and 5,5′-T-DAII showed good thermal stabilities with the 5% weight loss at 350 °C for 6,6′-T-DAII and at 332 °C for 5,5′-T-DAII (Fig. S2†). The melting points and crystallization points determined from the DSC profiles are 275 °C and 263 °C for 6,6′-T-DAII and 265 °C and 235 °C for 5,5′-T-DAII, respectively (Fig. S3†).
Single crystal structures
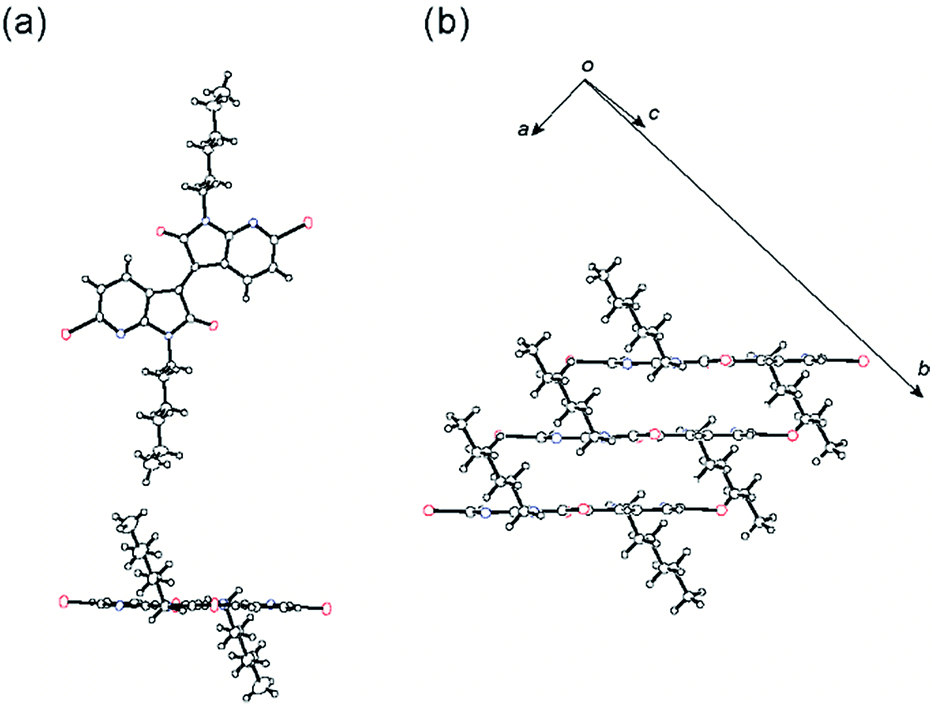
Suitable crystals of 6,6′-T-DAII, 5,5′-T-DAII, and 9a for the X-ray single crystal structure analysis were obtained by the slow diffusion of hexane into chloroform solutions of 6,6′-T-DAII and 5,5′-T-DAII and slow evaporation of the chloroform–hexane solution of 9a. The crystallographic data are listed in Table S1.† The molecules of 6,6′-T-DAII crystallize in the triclinic system with a space group P(![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) while molecule 9a and 5,5′-T-DAII belong to the monoclinic system with the space group P21/n for 9a and P21/c for 5,5′-T-DAII, respectively. Thereby, in the crystals, the half molecule for 6,6′-T-DAII, 9a, and 5,5′-T-DAII is crystallographically independent. As for the structure of 5,5′-T-DAII, two molecular stacks are orthogonally aligned to each other along the c axis. Molecule 9a has a flat π-framework and forms slipped one-dimensional stacks with the π–π stacking distance of 3.36 Å, demonstrating that this DAII moiety is promising to obtain the planarized platform as predicted from the geometry optimization (Fig. 3).
) while molecule 9a and 5,5′-T-DAII belong to the monoclinic system with the space group P21/n for 9a and P21/c for 5,5′-T-DAII, respectively. Thereby, in the crystals, the half molecule for 6,6′-T-DAII, 9a, and 5,5′-T-DAII is crystallographically independent. As for the structure of 5,5′-T-DAII, two molecular stacks are orthogonally aligned to each other along the c axis. Molecule 9a has a flat π-framework and forms slipped one-dimensional stacks with the π–π stacking distance of 3.36 Å, demonstrating that this DAII moiety is promising to obtain the planarized platform as predicted from the geometry optimization (Fig. 3).
 |
| | Fig. 3 (a) ORTEP plots at 50% ellipsoid probability and (b) molecular packing of 9a. | |
It is instructive to compare the molecular and packing structures of the two structural isomers of 6,6′-T-DAII and 5,5′-T-DAII, which would have a strong influence on the carrier-transport. Both π-frameworks adopt a perfectly planar geometry. The planarity of 6,6′-T-DAII well agrees with the optimized structures obtained from the calculations, while the planarized π-framework of 5,5′-T-DAII is inconsistent with the estimated distorted structure. This observation is ascribable to the fact that the intermolecular ordering predominantly determines the molecular geometry. Molecules 6,6′-T-DAII and 5,5′-T-DAII pack into the slipped one-dimensional columnar structures with interplanar distances of 3.51 Å for 6,6′-T-DAII and 3.32 Å for 5,5′-T-DAII (Fig. 4). For the analysis of 6,6′-T-DAII, we treated the terminal flanking thiophenes as a rotational disorder with S1 and C17 atoms, similarly to a previous report.42 Within the stack, the hexyl chain orientations are quite different; both hexyl chains of 6,6′-T-DAII are oriented in plane whereas those of 5,5′-T-DAII extend out of the DAII plane. These findings indicate that the planar π-framework is more important than the hexyl chain conformation in order to realize the co-facial segregated molecular packing. In the structure of 6,6′-T-DAII, the disordered flanking thiophene are, presumably, arising from the removal of steric hindrance around the bond connecting the central DAII unit and peripheral thiophenes. These findings imply that the DAII unit itself has a strong tendency to interact with each other to make a columnar structure that forms a charge-transport path, in which no noticeable conformational lock that is important to the planarize molecular backbone is observed. The calculated transfer integrals43 also indicate the one-dimensional transport of 6,6′-T-DAII and 5,5′-T-DAII, in which among the positionally disordered atoms of 6,6′-T-DAII, the S1 and C17 atoms bearing a larger occupancy are employed in the calculation; the HOMO transfer integrals within the stacks are t = 29 meV for 6,6′-T-DAII and t = 24 meV for 5,5′-T-DAII, and LUMO transfer integrals are t = 13 meV for 6,6′-T-DAII and t = 29 meV for 5,5′-T-DAII, while the other interstack transfers are negligible.
 |
| | Fig. 4 ORTEP plots at 50% ellipsoid probability and molecular packings of 6,6′-T-DAII (a and b) and 5,5′-T-DAII (c and d). | |
Optical properties
The UV-Vis-NIR absorption spectra were recorded in the solutions and thin films of 6,6′-T-DAII and 5,5′-T-DAII, and the data are summarized in Table 1. In solutions, the molecules exhibited two absorption bands, in which the high-energy bands accompanying with shoulders are centered at 326 nm for 6,6′-T-DAII and at 318 nm for 5,5′-T-DAII, and the low-energy bands are centered at 553 nm for 6,6′-T-DAII and 540 nm for 5,5′-T-DAII. The optical band gaps of 6,6′-T-DAII and 5,5′-T-DAII were determined from the onset of absorption to be 1.82 and 1.89 eV, respectively. When going from solutions to thin films, all the absorption profiles are red-shifted and become more structural, which might be due to the intermolecular interactions in the solid state. It is noteworthy that the molar absorption coefficients in the low-energy band showed an especially clear dependence in the patterns of substitution; the absorption intensity of 5,5′-T-DAII largely decreased in comparison to 6,6′-T-DAII. In order to further examine the optical transitions of 6,6′-T-DAII and 5,5′-T-DAII, we carried out TD-DFT calculations at the ωB97-XD/6-311++G(d,p) level (Table 2). The low-energy band showing a remarkable difference between 6,6′-T-DAII and 5,5′-T-DAII is assigned to the HOMO → LUMO transition with a mainly π–π* character. The oscillator strength for this transition was estimated to be 0.847 for 6,6′-T-DAII and 0.148 for 5,5′-T-DAII, well agreeing with the experimental observation. In spite of the large absorption difference, the optical bandgaps of 6,6′-T-DAII and 5,5′-T-DAII are almost similar at around 1.8 eV. Therefore, the substitution patterns in the DAII unit have a strong influence on the oscillator strength, but do not affect the optical bandgap as similarly reported for IID (Fig. 5).24
Table 1 Summary of electrochemical and optical properties
| |
E
LUMO
a (eV) |
E
HOMO
a (eV) |
λ
solmax(εmax)b (nm) (×103 M−1 cm−1) |
λ
filmmax
c (nm) |
E
optg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d (eV) d (eV) |
|
Estimated from the onset point of the reduction step and optical bandgaps.
Measured in CHCl3 solutions.
Spin-coated film from CHCl3 solutions.
Estimated from the solution absorption edge.
|
|
6,6′-T-DAII
|
−3.57 |
−5.39 |
326 (3.69), 553 (2.67) |
333, 637 |
1.82 |
|
5,5′-T-DAII
|
−3.64 |
−5.53 |
318 (4.96), 540 (0.26) |
320, 579 |
1.89 |
Table 2 DFT calculated optical transitionsa
| |
λ
max (eV) |
f
|
Main contributions to vertical transitions |
|
The transitions with oscillator strengths over 0.1 are listed.
|
|
6,6′-T-DAII
|
2.73 |
0.847 |
HOMO → LUMO (45%) |
| 4.13 |
0.856 |
HOMO−2 → LUMO (27%) |
| 4.38 |
0.496 |
HOMO → LUMO+2 (25%) |
|
5,5′-T-DAII
|
2.86 |
0.148 |
HOMO → LUMO (42%) |
| 3.70 |
0.110 |
HOMO−7 → LUMO (21%), HOMO−2 → LUMO (17%) |
| 4.03 |
0.380 |
HOMO−2 → LUMO (14%), HOMO−2 → LUMO (20%) |
| 4.50 |
1.273 |
HOMO−1 → LUMO+2 (16%), HOMO−9 → LUMO (25%) |
 |
| | Fig. 5 UV-Vis absorption spectra of 6,6′-T-DAII and 5,5′-T-DAII in (a) CHCl3 solutions and (b) thin films. | |
Electrochemical properties
The electrochemical properties were examined by cyclic voltammetry (CV) in a standard three electrode electrochemical cell with an argon saturated dichloromethane solution containing 0.1 M Bu4NPF6 at room temperature using Ag/AgNO3 as the reference electrode (Table 1). Both 6,6′-T-DAII and 5,5′-T-DAII exhibited irreversible reduction step (Fig. S4†). In the positive scan, no noticeable steps were observed, indicating the strong electron-accepting character of DAII unit. From the reduction onset potentials referenced to the Fc/Fc+ couple, in which the absolute energy level is assumed to be 4.8 eV under vacuum,44 the LUMO levels were estimated to be −3.57 eV for 6,6′-T-DAII and −3.64 eV for 5,5′-T-DAII. Based on the LUMO levels and optical bandgaps, the HOMO levels were estimated to be −5.39 eV for 6,6′-T-DAII and −5.53 eV for 5,5′-T-DAII. The similar LUMO level and deeper HOMO level of 5,5′-T-DAII relative to 6,6′-T-DAII are basically consistent with the trend of the DFT calculations.
Charge-transport properties
The charge-transporting properties of 6,6′-T-DAII and 5,5′-T-DAII were evaluated with a bottom-gate and top contact configuration in FET devices. The semiconducting layer was thermally deposited on a tetratetracontane (TTC) modified SiO2/Si substrate, and Au contacts were patterned by thermal evaporation. The TTC layer is known to form a superior defect-free interface with active layers to observe the intrinsic FET characteristics of the molecules.45 The measurement was performed in a vacuum to eliminate the influence of the ambient condition. The FET data are summarized in Table 3, and typical transfer curves are shown in Fig. 6. The devices made of 6,6′-T-DAII and 5,5′-T-DAII showed an ambipolar charge-transport, and their hole and electron mobilities are on the same order of 10−3 cm2 V−1 s−1. The electron mobility is slightly improved relative to the hole mobility, suggesting an n-type dominant character due to the electron-deficient DAII core. Most importantly, the devices made of 6,6′-T-DAII and 5,5′-T-DAII exhibited a comparable carrier-transport performance irrespective of the thiophene-flanked positions, suggesting that chemical modifications at both the 5- and 6-connections of the DAII core contribute to the carrier-transport. Of particular emphasis is that the 5,5′-substituted 5,5′-T-DAII obviously improves the carrier mobilities, especially concerning electron-transport, as compared to the 6,6′-substituted 6,6′-T-DAII. When considering the molecular packing in the crystal structures, this is ascribable to the smaller interplanar distance of 5,5′-T-DAII without the absence of any positional disorder of the flanked-thiophenes, thus realizing a more compact molecular packing within the stack. In general, the connection at the 5-position of the DAII core, namely, cross-conjugation, is considered to prevent lateral π-conjugation, implying negative impressions for making co-facial molecular π-stacks. However, we should state that the expanding backbone at the 5-position on the DAII core affords rather active influences on the charge-transport (Fig. 7).
| Compound |
p-type |
n-type |
|
µ
ave (cm2 V−1 cm−1) |
µ
max (cm2 V−1 cm−1) |
V
th (V) |
I
on/Ioff |
µ
ave (cm2 V−1 cm−1) |
µ
max (cm2 V−1 cm−1) |
V
th (V) |
I
on/Ioff |
|
Measurements were carried out under vacuum. Average mobilities of at least 12 devices are listed.
|
|
6,6′-T-DAII
|
1.0 × 10−3 |
2.1 × 10−3 |
−75 |
104 |
1.5 × 10−3 |
2.0 × 10−3 |
31 |
104 |
|
5,5′-T-DAII
|
1.7 × 10−3 |
2.6 × 10−3 |
−120 |
104 |
3.8 × 10−3 |
8.6 × 10−3 |
55 |
104 |
 |
| | Fig. 6 The typical transfer curves of (a) p-channel and (b) n-channel scans for 6,6′-T-DAII and (c) p-channel and (d) n-channel scans for 5,5′-T-DAII. | |
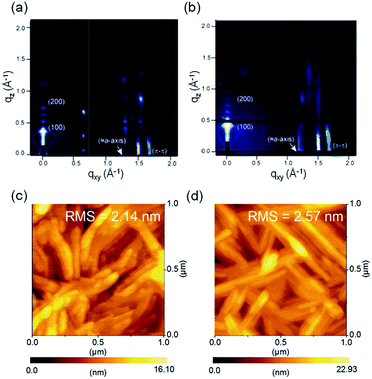 |
| | Fig. 7 GIXD patterns and AFM height images of (a) and (c) for 6,6′-T-DAII and (b) and (d) for 5,5′-T-DAII. | |
Thin film microstructure and morphologies
The X-ray diffraction (XRD) patterns and atomic force microscopy (AFM) images of the thin films of 6,6′-T-DAII and 5,5′-T-DAII were employed to examine the thin-film microstructures and surface morphologies. Thin films of 6,6′-T-DAII and 5,5′-T-DAII displayed sharp and intense primary peaks in the out-of-plane direction at the qz of 0.37 for 6,6′-T-DAII and at 0.41 for 5,5′-T-DAII, suggesting the presence of a periodic packing motif normal to the substrate, and the corresponding d-spacings are 17.2 Å for 6,6′-T-DAII and 15.2 Å for 5,5′-T-DAII. In the in-plane-direction, 6,6′-T-DAII and 5,5′-T-DAII displayed peaks corresponding to the π–π stacking distances of 3.75 Å as well. Additionally, the peaks of 6,6′-T-DAII and 5,5′-T-DAII centered at the q−1 of 1.25 corresponding to ≈5.0 Å, which is approximately consistent with the lengths of the crystallographic a axis in the single crystal structures of 6,6′-T-DAII and 5,5′-T-DAII. Although it is hard to determine the exact molecular orientation, when considering the molecular lengths of 6,6′-T-DAII and 5,5′-T-DAII estimated from X-ray single crystal structure analyses, these observations indicate that uniformly stacked columnar structures are organized parallel to the substrate, in which the molecular long axes of 6,6′-T-DAII and 5,5′-T-DAII are tilted by ≈20° normal to the substrate. These parallel molecular orders of the carrier-transporting plane are preferable for carrier transport in FETs. In the AFM height images, both thin films were composed of almost the same interconnected fibril-like grains. These thin-film microstructures and surface morphologies are well rationalized with their comparable FET performances. It is interesting that the structural isomers 6,6′-T-DAII and 5,5′-T-DAII, which possess different platforms and electronic structures originating from the substitution patterns, exhibited the same order of magnitude of carrier mobilities, and 5,5′-T-DAII displayed further enhanced carrier mobilities. These results reveal that the present DAII core does not reduce the charge-transport properties irrespective of the 5- or 6-substitution patterns.
Conclusions
In conclusion, a novel electron-deficient building unit, DAII, was designed and synthesized. The dibrominated-DAIIs are useful coupling partners with a synthetic diversity for preparing semiconducting materials. The optical, electrochemical, and carrier-transport properties were examined by specifically on the impact of the connection patterns on the structure–property relationship. The two structural isomers of 6,6′-T-DAII and 5,5′-T-DAII exhibited an ambipolar semiconductor performance with hole and electron carrier mobilities of 10−3 cm2 V−1 s−1. Of particular interest is that the 5,5′-substitution on the DAII core improved carrier-transport characteristics in comparison to the 6,6′-substitution. This finding reveals that the molecular packing coming from the connection pattern is an important factor to govern the carrier-transport even if the cross-conjugated FMOs exist on the backbone, which is generally considered to result in poor intermolecular couplings. Our preliminary results demonstrated that the DAII unit is a promising n-type building block and provides an option of connection patterns for creating small-molecule and polymeric semiconductors. The exploration of new organic semiconductors composed of the DAII unit is currently under way.
Acknowledgements
This study was partly supported by Grant-in-Aid for Scientific Research (C) (No. 26410087) from the Ministry of Education, Culture, Sports, Science and Technology (for M. A.). The authors are grateful to the Center for Advanced Materials, Tokyo Institute of Technology, for the microanalysis. The authors also thank Hiroyasu Sato and Rigaku Corporation for X-ray crystal structure analyses. GIWAX experiments were performed on BL45XU at SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2015B1105 and 2015B1690). The numerical calculations were carried out on the TSUBAME2.5 supercomputer at the Tokyo Institute of Technology, Tokyo, Japan, and on the supercomputer at the Research Center for Computational Science, Okazaki, Japan.
Notes and references
- A. R. Murphy and J. M. J. Frechet, Chem. Rev., 2007, 107, 1066–1096 CrossRef CAS PubMed.
- J. M. Anthony, A. Faccetti, M. Heeny, S. R. Marder and X. W. Zhan, Adv. Mater., 2010, 22, 3876–3892 CrossRef CAS PubMed.
- J. Zaumseil and H. Srringhaus, Chem. Rev., 2007, 107, 1296–1323 CrossRef CAS PubMed.
- A. C. Arias, J. D. MacKenzie, I. McCulloch, J. Rivnay and A. Salleo, Chem. Rev., 2010, 110, 3–24 CrossRef CAS PubMed.
- J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724–6746 CrossRef CAS PubMed.
- A. Facchetti, Mater. Today, 2013, 16, 123–132 CrossRef CAS.
- J. Li, Y. Zhao, H. S. Tan, Y. Guo, C. A. Di, G. Yu, Y. Liu, M. Lin, S. H. Lim, Y. Zhou, H. Su and B. S. Ong, Sci. Rep., 2012, 2, 754 Search PubMed.
- I. Kang, H. J. Yun, D. S. Chung, S. K. Kwon and Y. H. Kim, J. Am. Chem. Soc., 2013, 135, 14896–14899 CrossRef CAS PubMed.
- G. Kim, S. J. Kang, G. D. Dutta, Y. K. Han, T. J. Shin, Y. Y. Noh and C. Yang, J. Am. Chem. Soc., 2014, 136, 9477–9483 CrossRef CAS PubMed.
- B. Kang, R. Kim, S. B. Lee, S. K. Kwon, Y. H. Kim and K. Cho, J. Am. Chem. Soc., 2016, 138, 3679–3686 CrossRef CAS PubMed.
- H. J. Yun, S. J. Kang, Y. Xu, S. O. Kim, Y. H. Kim, Y. Y. Noh and S. K. Kwon, Adv. Mater, 2014, 26, 2636–2642 CrossRef PubMed.
- J. E. Anthony, A. Faccetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater, 2010, 22, 3876–3892 CrossRef CAS PubMed.
- X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef CAS PubMed.
- Z. H. Chen, Y. Zheng, H. Han and A. Faccetti, J. Am. Chem. Soc., 2009, 131, 8–9 CrossRef CAS PubMed.
- C. Tozlu, S. Erten-Ela, T. B. Singh, N. S. Sariciftci and S. İcli, Synth. Met., 2013, 172, 5–10 CrossRef CAS.
- J. Lee, A. R. Han, H. Yu, T. J. Shin, C. Yang and J. H. Oh, J. Am. Chem. Soc., 2013, 135, 9540–9547 CrossRef CAS PubMed.
- I. Kang, H. J. Yun, D. S. Chung, S. K. Kwon and Y. H. Kim, J. Am. Chem. Soc., 2013, 135, 14896–14899 CrossRef CAS PubMed.
- Y. Kanbur, M. Irimia-Vladu, E. D. Glowacki, G. Voss, M. Baumgartner, G. Schwödiauer, H. Sitter, Z. Kücükyavuz, S. Bauer and N. S. Sariciftci, Org. Electron., 2012, 13, 919–924 CrossRef CAS PubMed.
- J. Mei, D. H. Kim, A. L. Ayzner, M. F. Toney and Z. Bao, J. Am. Chem. Soc., 2011, 133, 20130–20133 CrossRef CAS PubMed.
- T. Lei, Y. Gao, X. Zhou, Y. Peng, J. Bian and J. Pei, Chem. Mater., 2012, 24, 1762–1770 CrossRef CAS.
- T. Lei, Y. Cao, Y. Fan, C. J. Liu, S. C. Yuan and J. Pei, J. Am. Chem. Soc., 2011, 133, 6099–6101 CrossRef CAS PubMed.
- R. Stalder, J. Mei, K. R. Graham, L. A. Estrada and J. R. Reynolds, Chem. Matter., 2014, 26, 664–678 CrossRef CAS.
- Y. Ren, A. M. Hiszpanski, L. W. Brooks and Y. L. Loo, ACS Appl. Mater. Interfaces, 2014, 6, 14533–14542 CAS.
- G. W. P. V. Pruissen, J. Brebels, K. H. Hendriks, M. M. Wienk and R. A. J. Janssen, Macromolecules, 2015, 48, 2435–2443 CrossRef.
- L. A. Estrada, R. Stalder, K. A. Abboud, C. Risko, J. L. Brédas and J. R. Reynolds, Macromolecules, 2013, 46, 8832–8844 CrossRef CAS.
- G. W. P. Van Pruissen, F. Gholamrezaie, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem., 2012, 22, 20387–20393 RSC.
- R. S. Ashraf, A. J. Knonemeijer, D. I. James, H. Sirringhaus and I. McCulloch, Chem. Commun., 2012, 48, 3939–3941 RSC.
- L. Meager, M. Nikolka, B. C. Schroeder, C. B. Nielsen, M. Planell, H. Bronstein, J. W. Rumer, D. L. James, R. S. Ashraf, A. Sadhanala, P. Hayoz, J. C. Flores, H. Sirringhaus and I. McCulloch, Adv. Funct. Mater., 2014, 24, 7109–7115 Search PubMed.
- T. Odajima, M. Ashizawa, Y. Konosu, H. Matsumoto and T. Mori, J. Mater. Chem. C, 2014, 2, 10455–10467 RSC.
- T. Hasegawa, M. Ashizawa and H. Matsumoto, RSC Adv., 2015, 5, 61035–61043 RSC.
- T. Hasegawa, M. Ashizawa, J. HIyoshi, S. Kawauchi, J. Mei, Z. Bao and H. Matsumoto, Polym. Chem., 2016, 7, 1181–1190 RSC.
- G. Kim, S. Kang, G. K. Dutta, Y. Han, T. J. Shin, Y. Noh and C. Yang, J. Am. Chem. Soc., 2014, 136, 9477–9483 CrossRef CAS PubMed.
- M. Ashizawa, N. Masuda, T. Higashino, T. Kadoya, T. Kawamoto, H. Matsumoto and T. Mori, Org. Electron., 2016, 35, 95–100 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, The molecular orbital calculation was carried out using the Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- J. Huang, Z. Mao, Z. Chen, D. Gao, C. Wei, W. Zhang and G. Yu, Chem. Mater., 2016, 4, 1208–1214 Search PubMed.
- G. D. Miguel, L. Camacho and E. M. García-Frutos, J. Mater. Chem. C, 2016, 4, 1208–1214 RSC.
- S. Minakata, M. Komatsu and Y. Ohshiro, Synthesis, 1992, 661–663 CrossRef CAS.
- X. K. Wee, T. Yang and M. L. Go, ChemMedChem, 2012, 777–791 CrossRef CAS PubMed.
- P. W. Wu, Y. M. Cheng, W. T. Hsieh, Y. H. Wang, C. Y. Wei and P. T. Chou, ChemMedChem, 2007, 1071–1075 CrossRef CAS PubMed.
- X. Cheng, K. H. Merz, S. Vatter, J. Christ and S. Wölfi, Bioorg. Med. Chem., 2014, 247–255 CrossRef CAS PubMed.
- J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508–524 CrossRef.
- J. Chisaka, M. Lu, S. Nagamatsu, M. Chikamatsu, Y. Yoshida, M. Goto, R. Azumi, M. Yamashita and K. Yase, Chem. Mater., 2007, 19, 2694–2701 CrossRef CAS.
- T. Mori, A. Kobayashi, Y. Sasaki, H. Kobayashi, G. Saito and H. Inokuchi, Bull. Chem. Soc. Jpn., 1984, 57, 627–633 CrossRef CAS.
- B. W. D'Andrade, S. Datta, S. R. Forrest, P. Djurovich, E. Polikarpov and M. E. Thompson, Org. Electron., 2005, 6, 11–20 CrossRef.
- M. Irimia-Vlada, E. D. Glowacki, P. A. Troshin, G. Schwabegger, L. Leconat, D. K. Susarova, O. Krystal, M. Ullah, Y. Kanbur, M. A. Bodea, V. F. Razumov, H. Sitter, S. Bauer and N. S. Sariciftci, Adv. Mater., 2012, 24, 375–380 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information on material synthesis, DFT calculations, thermal properties, redox properties, X-ray single crystal structure analysis, and fabrication and characterization of OFETs. CCDC 1479803–1479805. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra17424h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a