
Highly sensitive determination of ethyl carbamate in alcoholic beverages by surface-enhanced Raman spectroscopy combined with a molecular imprinting polymer†
Abstract
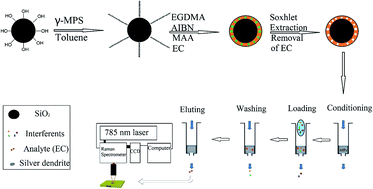
A simple and reliable method for fast extraction and sensitive detection of ethyl carbamate (EC) in rice wine and fruit brandy based on the integration of molecularly imprinted polymers and surface-enhanced Raman spectroscopy (MIPs–SERS) was developed. Molecularly imprinted polymer microspheres were synthesized as artificial antibodies towards EC. Adsorption tests and thermogravimetric analysis validated the specific selectivity and stability of MIPs respectively. The synthesized MIPs was used as sorbents in solid-phase extraction (SPE) and could selectively separate and enrich EC from rice wine and fruit brandy samples with little interference. A silver dendrite nanostructure was employed as a SERS-active substrate for the enhancement of Raman signals. Spectra processed by principal component analysis (PCA) can clearly differentiate Raman signatures of wine samples with various EC contents. A partial least square (PLS) regression model demonstrated a good fitting effect (the correlation coefficient of calibration and prediction were 0.9614 and 0.9456, and 0.9559 and 0.9393 respectively for rice wine and fruit brandy) between the predicted and the reference data of EC in rice wine and fruit brandy samples. Our results show that the high selectivity of MIPs and the fingerprint Raman identification can be integrated into a novel nano-biosensor for fast and efficient detection of hazardous substances (EC) in complex rice wine and fruit brandy samples.
 Please wait while we load your content...
Please wait while we load your content...

