
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Co-adsorption of O2 and H2O on Al(111) surface: a vdW-DFT study
Xin
Wei
a,
Chaofang
Dong
*a,
Zhanghua
Chen
b,
Kui
Xiao
a and
Xiaogang
Li
a
aCorrosion and Protection Center, Key Laboratory for Corrosion and Protection (MOE), University of Science and Technology Beijing, Beijing 100083, China. E-mail: cfdong@ustb.edu.cn; Fax: +86-10-62334005; Tel: +86-10-62333931-518
bSchool of Mathematics and Physics, University of Science and Technology Beijing, Beijing, 100083, China
First published on 17th August 2016
Abstract
Using first-principles calculations based on van der Waals density functional theory, we systematically studied the co-adsorption behavior of H2O and O2 on Al(111) surfaces. The study consists of two parts: (i) the adsorption of H2O molecules on O pre-adsorbed Al(111) surfaces, and (ii) co-adsorption of H2O and O2 molecules on a clean Al(111) surface. H2O adsorbs on O pre-adsorbed Al(111) surfaces in the form of a molecule, while both the adsorption energies and partial density states results prove that the adsorption of H2O is strengthened with the increasing pre-adsorbed O coverage. An Al atom bonded with H2O is pulled out of the surface in all adsorbed structures because of the repulsion between pre-adsorbed O atoms and the O atoms of H2O. For the co-adsorption of H2O and O2 on a clean Al(111) surface, H2O molecules can dissociate into OH and H when both of the two O atoms of O2 can interact with H atoms of H2O, or else they will adsorb on the surface keeping the behavior of single adsorption. For the dissociation adsorption of H2O, the redistribution of charge occurs and the value of charge transfer from the surface and O2 to H2O is much larger than that of molecular adsorption, which is larger than 0.1.
1. Introduction
The oxidation of Al (by O2 or H2O) is one of the most thoroughly studied reactions1–15 because of the importance of this chemical interaction in electrochemistry, heterogeneous catalysis, and corrosion. Studies concerning the chemisorption and dissociation of O2 and H2O molecules are of major significance because they are the fundamental steps for the oxidation of Al which is an important process anywhere Al is used. The mechanisms of O2 molecule chemisorption and dissociation on Al surfaces have been studied using a large variety of experimental and theoretical techniques.1–10 Experiments and theoretical calculations support chemisorption only on the fcc site of the first layer of Al(111),7,9 and O2 does not penetrate into subsurface sites.10 Brune et al.7 studied the adsorption behavior of O2 on the Al(111) surface by using a scanning tunneling microscope, they found that O2 is far from steady under room temperature and dissociation O atoms are adsorbed with monatomic shape. The interaction of H2O with metal has been the subject of numerous surface science investigations.11–14 Some of these studies can develop a general description of the structure, bonding and reactivity of H2O with clean and O pre-adsorbed surfaces.4,5 Netzer and Madey's study indicates that H2O adsorbed on clean Al(111) trend to O atom close to surface and H atoms away from surface.15 At 80 K, they also found that H2O is adsorbed predominantly in molecular form, and bonding occurs through the O atom, with the molecular axis tilted away from the surface normal.Co-adsorption behaviors of gas molecules on metal surfaces have been subject to extensive studies.16–21 However, little work has been done on the co-adsorption behavior of O2 and H2O on Al surface, which is of significance for the passive film forming and corrosion resistance.22,23 Only a few studies concerning the adsorption of H2O on O pre-adsorbed Al surface have been investigated.12–14 J. E. Crowell et al.13 investigated the adsorption of H2O on both clean and O pre-adsorbed Al(111) surface by vibrational spectroscopy using electron energy loss spectroscopy. On the clean surface, adsorption is predominantly molecular, while in the presence of oxygen, adsorption is predominantly dissociative. Besides, the production of adsorbed hydroxyl species from H2O reaches a maximum at 250 K on the clean surface and at 350 K on the O pre-adsorbed surface. The hydroxyl species decompose above these temperatures to evolve hydrogen and further oxidize the Al(111) surface. Guo et al.14 symmetrically studied the adsorption behavior of a single molecular H2O on a clean and an O pre-adsorbed Al(111) surface, and also its corresponding dissociation reactions using first-principle method based on DFT. The results also showed that the adsorption O can promote the dehydrogenation of H2O.
We have already studied the single adsorption of O2 and H2O on clean Al(111) surface and the effect of pre-adsorbed O atoms on the adsorption of H2O in our previous work.24 In this study, we have utilized first-principles method based on van der Waals density functional density (vdW-DFT)25 to examine the co-adsorption behavior of O2 and H2O. The adsorption of H2O on O pre-adsorbed Al(111) surfaces with different O coverage is further calculated based on previous study to investigate the effect of the coverage of pre-adsorbed O atoms on the dissociation of H2O. The co-adsorption of O2 and H2O on Al(111) surface is investigated initiated from eight different configurations. We study the structures and electronic properties of all calculated configurations to investigate the oxidation of Al by O2 and H2O.
2. Computational details
All calculations presented in this work are carried out utilizing MedeA-VASP 5.4 software,26,27 which is a fast and highly reliable electronic structure method based on DFT.28 The calculation is conducted in a plane-wave basis, using the projector-augmented wave method.29 According our previous results,24 all calculations only involving O atoms were performed by standard DFT method with a spin polarization, which is a calculation based on the GGA-PBE exchange-correlation functional for describing the interactions. The vdW-DFT taking into account in an approximate manner the dispersive forces and van der Waals interactions are used for the adsorption of H2O molecule co-adsorption of H2O and O2. The optimized van der Waals functional based on the Becke 86 (optB86-vdW30,31) is chose as the exchange functional, which trend to exhibit smallest errors for most systems investigated. The non-local vdW correlation was not defined for a spin polarized system. This approach does not rely on empirical force field and is true first principles technique, which is a strict algorithm. OptB86-vdW was mostly used to treat van der Waals type of systems, achieving quite good success.32–34The adsorption calculations are conducted on 6-layer slabs of Al(111) with a 12 Å vacuum gap. A (3 × 3) mesh is used for the adsorption calculation of a single H2O molecule on O pre-adsorbed surface and a (4 × 4) supercell is used to co-adsorption. The adsorbates and the three uppermost surface layers are allowed to move freely, and the bottom three layers are fixed. The electronic iterations convergence is 10−5 eV using the normal (blocked Davidson) algorithm. Periodic boundary conditions are set, leading to an infinite periodic system. The slab models are calculated with a (4 × 4 × 1) and (3 × 3 × 1) Monkhorst-Pack grid35 for two size supercells, respectively. All calculations are performed using a 520 eV cut off energy. The post-processing of results based on structure and charge density is constructed by VESTA.36
The so-called Bader scheme for dividing slab model into atomic regions has been used,37 which proved to be robust and efficient. Included in MedeA-VASP as a property module, this approach is based on a classic algorithm proposed by Bader, as implemented by Henkelman et al.38 The surface work function is the energy to extract one electron from surface to vacuum, which is calculated to characterize the surface electronic state. The property of partial density of states (PDOS) is calculated to investigate the interaction among H2O molecule with surfaces. The adsorption energy (Ead) values are calculated from the following expression:
| Ead = Eads + Esub − Eads/sub | (1) |
3. Results and discussions
3.1 Structures of O(fcc) pre-adsorbed Al(111) surface
O2 can adsorb on multiple original positions on Al(111) surface. Dissociated O atoms can stable adsorbed at fcc sites, which is investigated by both theoretical and experimental studies.39–41 The optimized structures and electronic properties of the adsorption of O atoms on fcc sites are investigated in this section. O atoms are arranged at fcc sites in the order of Fig. 1, corresponding to the coverage from 1/9 ML to 1 ML. Geometrical parameters, surface work function, adsorption energies and charge transfer from surface to each O atom are listed in Table 1. Considering the computational error, the charges transfer from Al atoms to each O atom is around 1.7e and independent on the O coverage, which is also demonstrated by the similar Al–O distance at around 1.8 Å. The average distances of Al–O in all adsorbed structures are all smaller than the radius sum of Al3+ and O2− (ref. 14) indicating a stronger interaction between Al and O by the form of chemical bond. The adsorption energies referenced O atoms are basically consistent with previous results.41 The average adsorption energy of each O atom increases with the increasing O coverage from 1/9 ML to 1 ML according to the results in Table 1. The electronic property of surface can be characterized by the parameter of work function which increases with the increasing O coverage (Table 1) indicating that the ability in offering electrons of surface is weakened due to the adsorption of O atoms, which will reflect its adsorption behavior. The adsorbed O atoms will capture electrons of Al atoms and this ability will strengthen with the increasing O coverage, which result in the surface work function increases.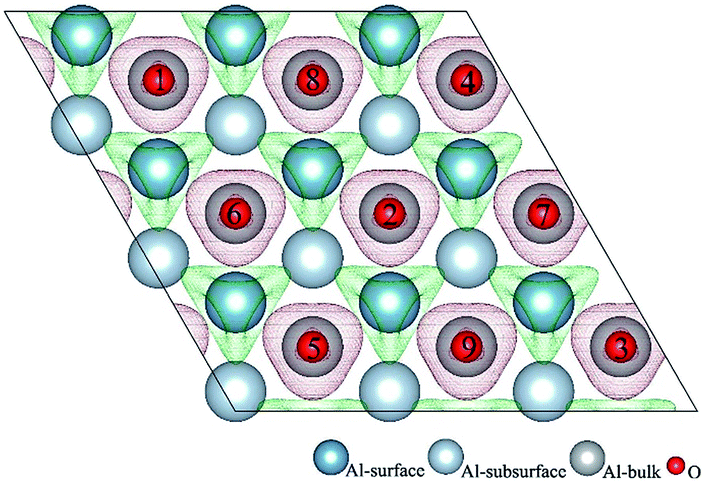 | ||
| Fig. 1 The difference charge density of O(fcc) atoms pre-adsorbed Al(111) surface. The 1–9 is the occupied order of O atoms. Red parts indicate a behaviour of accepting electrons and green parts indicate a behaviour of losing electrons. | ||
| O coverage/ML | Distance/Å Al–O (average) | Work function/eV | Charge transfer (Al → O) | Adsorption energy/eV |
|---|---|---|---|---|
| 0 | — | 4.088 | — | — |
| 1/9 | 1.87 | 4.085 | 1.72 | 7.53 |
| 2/9 | 1.86 | 4.093 | 1.73 | 7.55 |
| 3/9 | 1.85 | 4.126 | 1.73 | 7.55 |
| 4/9 | 1.84 | 4.156 | 1.71 | 7.71 |
| 5/9 | 1.84 | 4.200 | 1.70 | 7.83 |
| 6/9 | 1.83 | 4.265 | 1.70 | 7.90 |
| 7/9 | 1.82 | 4.334 | 1.70 | 7.97 |
| 8/9 | 1.82 | 4.404 | 1.68 | 8.01 |
| 1 | 1.80 | 4.476 | 1.68 | 8.07 |
3.2 Adsorbed structures of H2O on clean and O(fcc) pre-adsorbed Al(111) surfaces
We have studied the adsorption of a single H2O molecule on the clean and O(fcc) pre-adsorbed Al(111) surfaces. The adsorbed structures are shown in Fig. 2 and 3. The geometrical parameters, adsorption energies and charge transfer are listed in Table 2. As previous studies,14,15 the results shows that H2O adsorbed on clean Al(111) surface tends to O atom close to surface and H atoms away from surface, and bonding occurs through the O atom, with the molecular plane titled away from the surface normal. As similar as the clean Al(111), H2O adsorbs preferentially at the Al top site of the O(fcc) pre-adsorbed surfaces. Al atom bonded with H2O is pulled out of the surface with the increasing O coverage accompanied by the surface reconstruction. The distance of Al–O decreases from 2.17 Å to 1.93 Å with the increasing O coverage (Fig. 3). The distances are basically equal to the sum of O2− radius and Al3+ radius when the O coverage is equal or greater than 7/9 ML indicating a stronger chemical interaction. The deformation of H2O is also investigated. The average distance of O–H is 0.98 Å in all structures which is a little larger than that in gas and the ∠HOH tends to slightly increase with the increasing O coverage. Based on the results in Table 2, the adsorption energies of H2O on O(fcc) pre-adsorbed Al(111) surfaces did not monotonically increase with the increasing the O coverage. We infer that the effect of pre-adsorbed O atoms on H2O adsorption is dependent on the distance between the pre-adsorbed O and the adsorbed Al site by H2O. At the coverage of 1/9–3/9 ML, H2O adsorption energy is similar to that on the clean surface, due to the fact that pre-adsorbed O does not form bonds with the adsorbed Al site by H2O, while the adsorption energy of H2O is increased obviously when the O coverage increase to 1 ML, due to the strong interaction between pre-adsorbed O atoms and the adsorbed Al site by H2O. It is noteworthy that the direction of charge transfer changes at the O coverage interval between 7/9 ML and 8/9 ML. The ability of accepting electron of surface is stronger than that of H2O molecule when the O coverage is greater than the critical value. The adsorption energies are small at the low O coverage while it increases greatly when the O coverage is larger than 7/9 ML, which indicates that H2O prefer to interact with O pre-adsorbed Al(111) surface by losing electrons. Fig. 2 shows the relationship between surface work function of O pre-adsorbed Al(111) surfaces and charge transfer from surface to H2O. With the increasing O coverage, the charge transfer decreases with the increasing surface work function of O(fcc) pre-adsorbed Al(111) surfaces.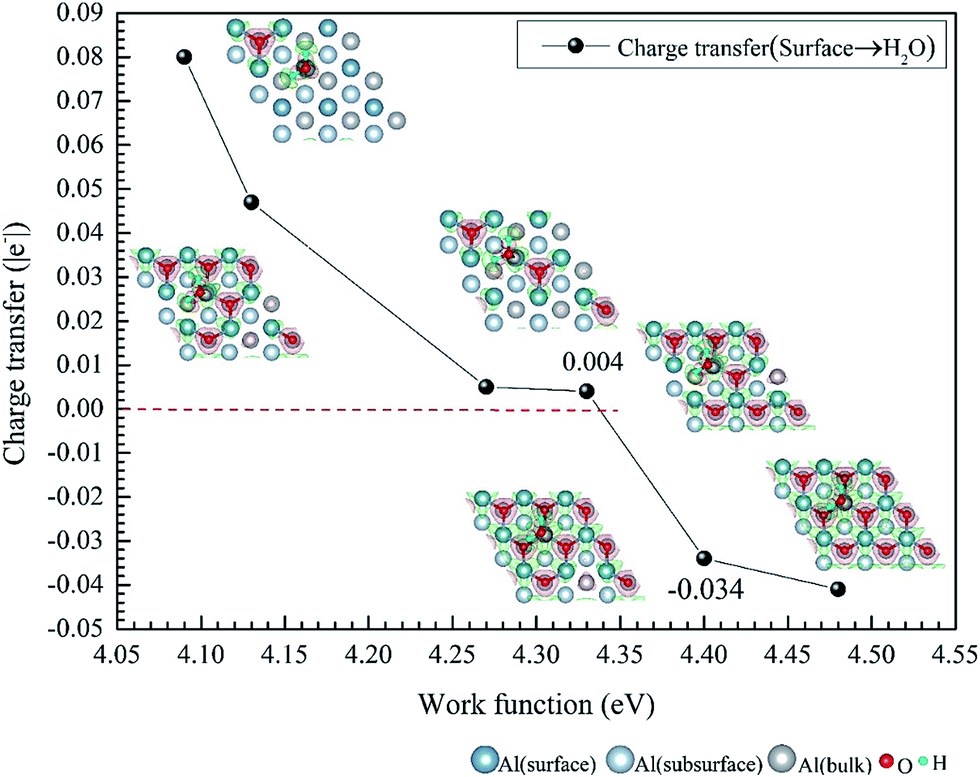 | ||
| Fig. 2 The relationship between surface work function of O(fcc) pre-adsorbed Al(111) surfaces and charge transfer from surface to H2O. The top view configurations are the difference charge density of adsorbed structures. | ||
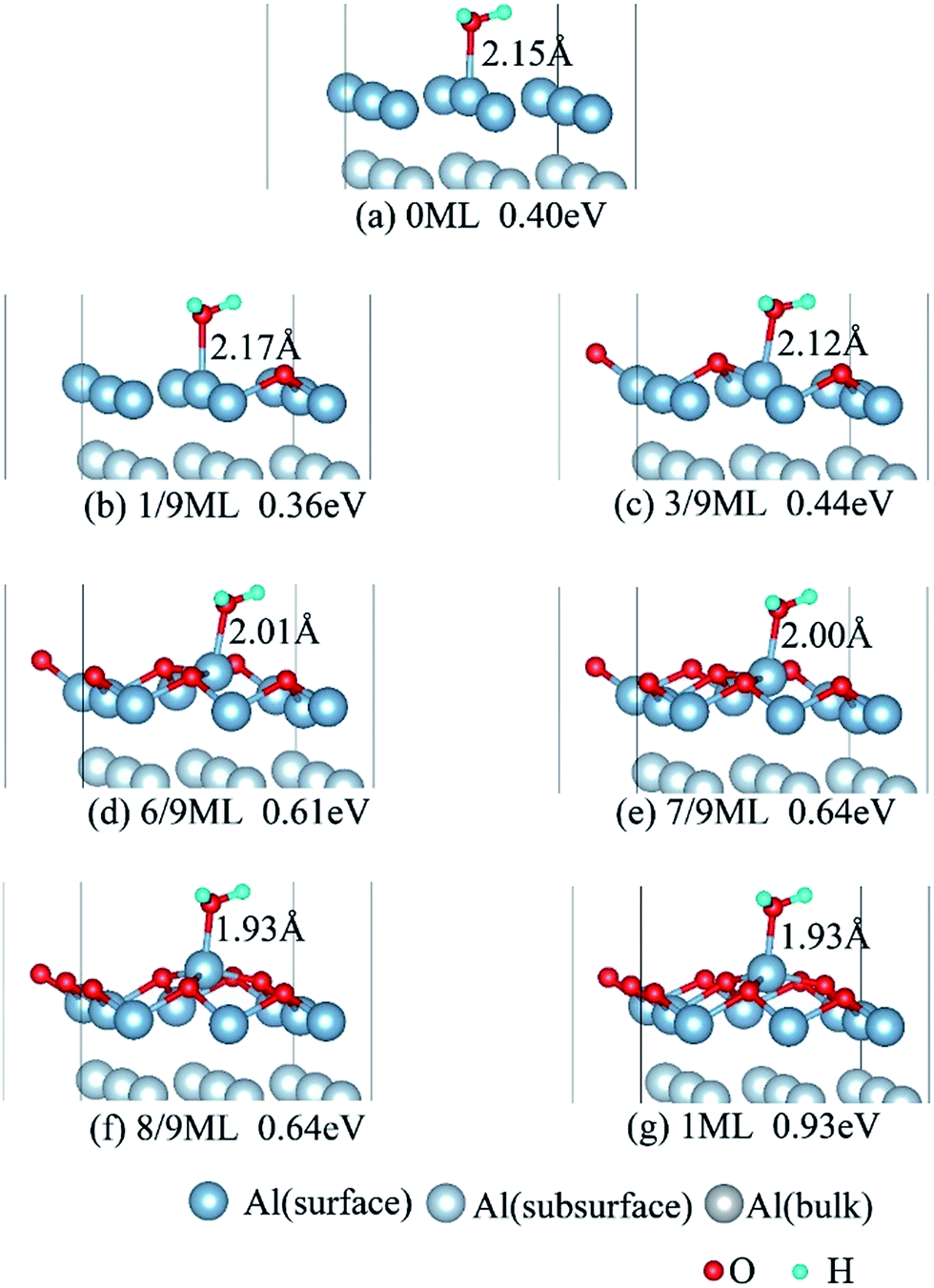 | ||
| Fig. 3 The side view of adsorbed structures of H2O on O(fcc) pre-adsorbed Al(111) surfaces. | ||
| O coverage/ML | Distance/Å | Angle ∠HOH/° | Adsorption energy/eV | Charge transfer (surf. → H2O) | |
|---|---|---|---|---|---|
| Al–O(H2O) | H–O | ||||
| 0 | 2.15 | 0.98 | 106.5 | 0.40 | 0.081 |
| 1/9 | 2.17 | 0.98 | 107.3 | 0.36 | 0.080 |
| 3/9 | 2.12 | 0.98 | 106.0 | 0.44 | 0.047 |
| 6/9 | 2.01 | 0.98 | 107.8 | 0.61 | 0.005 |
| 7/9 | 2.00 | 0.98 | 108.1 | 0.64 | 0.004 |
| 8/9 | 1.93 | 0.98 | 108.4 | 0.94 | −0.034 |
| 9/9 | 1.93 | 0.98 | 108.4 | 0.93 | −0.414 |
To better understand the interaction of the H2O molecule with the surface Al atoms, we calculated the PDOS of p orbital of O atom in H2O molecule and Al atom interacted with H2O molecule directly. The results are shown in Fig. 4. As can be seen, Fig. 4a–c shows the resonance of O(p) peak of H2O molecule and Al(p) peak of surface at the O coverage of 3/9, 6/9 and 1 ML, which indicates that there is an obvious interaction between them. Fig. 4d is the PDOS of O(p) of H2O adsorbed on the surface with O coverage of 3/9, 6/9 and 1 ML and H2O molecule in gas. The downshift of O(p) state with respect to the gas is characterized by the displacement of the peak. The result illustrates that the ranking of the adsorption strength of H2O on surface is O(fcc) pre-adsorbed surface (1 ML) > O(fcc) pre-adsorbed surface (6/9 ML) > O(fcc) pre-adsorbed surface (3/9 ML) > clean surface. We infer that surface with high O coverage favor the H2O–Al(111) surface interaction which agrees with the previous results of geometrical parameters and adsorption energies.
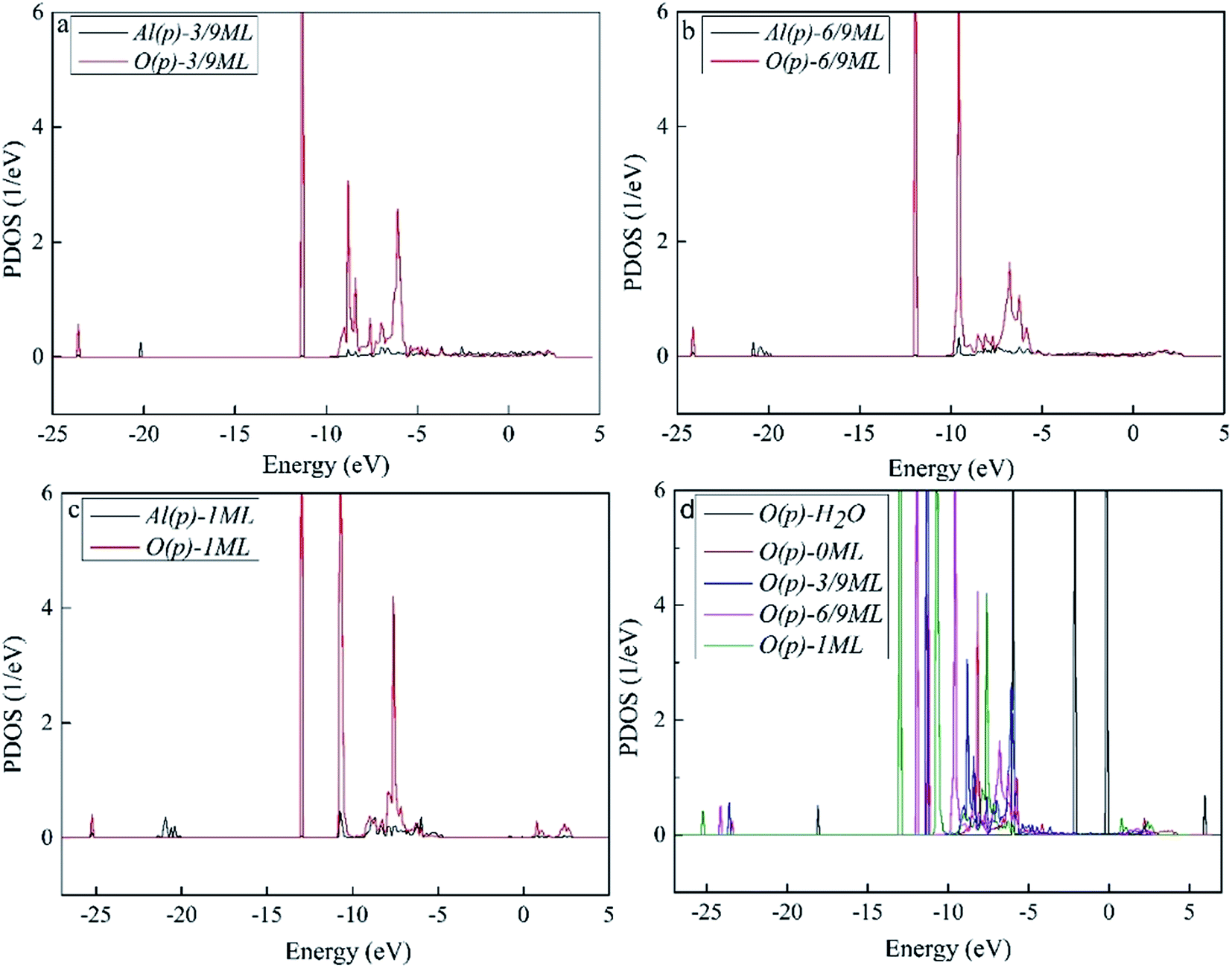 | ||
| Fig. 4 The PDOS results of (a) Al(p) and O(p) of H2O adsorbed Al(111) surface at the preadsorbed O coverage of 3/9 ML, (b) Al(p) and O(p) of H2O adsorbed Al(111) surface at the preadsorbed O coverage of 6/9 ML, (c) Al(p) and O(p) of H2O adsorbed Al(111) surface at the preadsorbed O coverage of 1 ML, and (d) O(p) of H2O at the preadsorbed O coverage of 0, 3/9, 6/9 and 1 ML and H2O in gas. | ||
3.3 Co-adsorption of H2O and O2 molecule on clean Al(111) surface
The result of section 3.2 illustrates that the pre-adsorbed O atoms can promote the deformation of H2O and strengthened its adsorption on Al(111) surface with the increasing O coverage. The transition states search results of our previous study24 and the work of Guo et al.14 also proved that the existence of pre-adsorbed O atom can greatly reduce the dissociation energy of H2O. In this section, the effect of O2 molecule on the dissociation and adsorption of H2O on Al(111) surface is investigated. Eight initial structures are calculated to investigate the co-adsorption behavior, which are marked from Co-1 to Co-8. The adsorbed structures can be classified into two groups which are the dissociated and molecular adsorption of H2O on Al(111) surface, which are listed in Tables 3 and 4, respectively. O2 molecule dissociates into O atoms and adsorbs on surface fcc or hcp sites in all structures, while H2O dissociates into OH and H only from Co-1, Co-2 and Co-3 structure, and adsorbs on surface top site by the form of molecule in the adsorbed structures of Co-4, 5, 6, 7, and 8. For the dissociated structures of H2O, OH adsorbs on top site and H bonds with O atom dissociated from O2 to produce OH which adsorbs on bridge site. For the adsorbed structure of Co-5, one of O atoms dissociated from O2 is permeated into subsurface, which is resulted by the co-adsorption of H2O. The geometrical parameters, adsorption energies and charge transfer from surface and O2 to H2O of the co-adsorbed structures of H2O and O2 on clean Al(111) surface are listed in Table 5. Considering the distance between Al and O of OH and H2O, the binding force of Al–OH is stronger than that of Al–H2O. The bond of O–H is elongated in OH corresponding to H2O molecule and there is an obvious deformation of H2O related the angle of ∠HOH.| Initial structure | Final structure | ||
|---|---|---|---|
| Top view | Top view | Side view | |
| Co-1 |
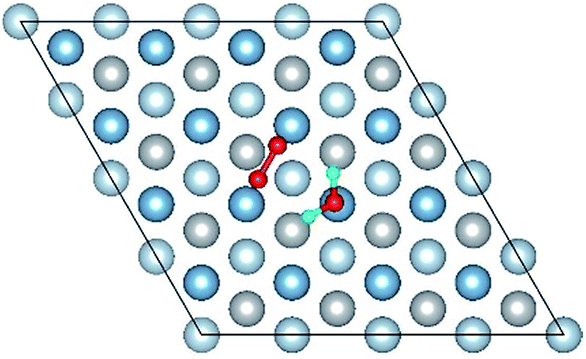
|
|
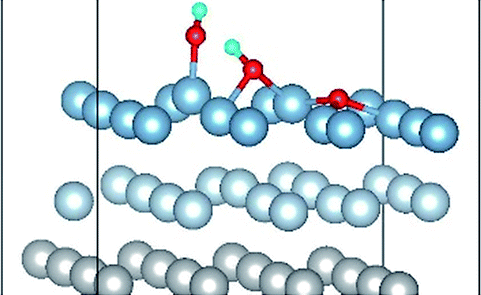
|
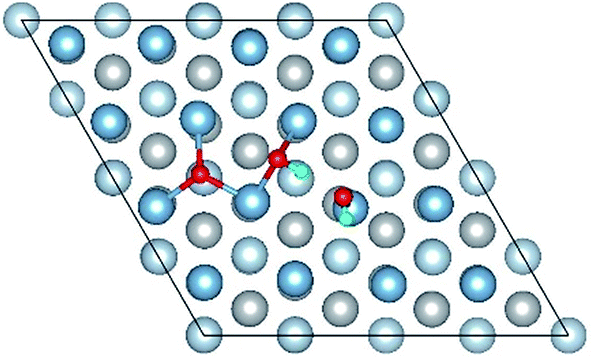
| Co-2 |
|
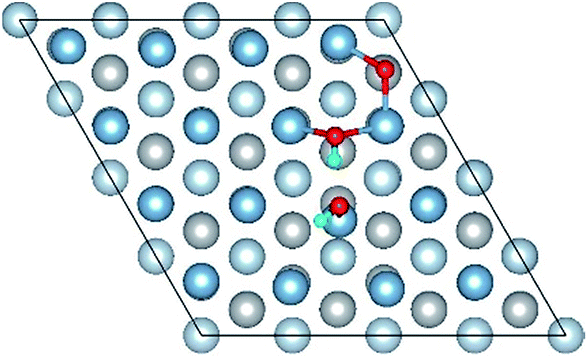
|
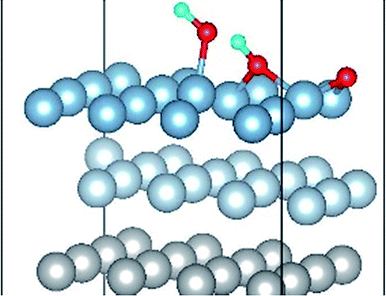
|
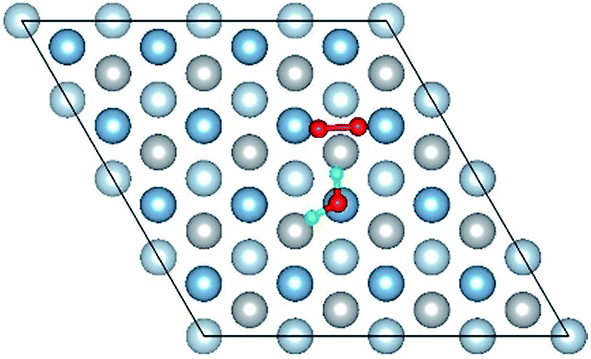
| Co-3 |
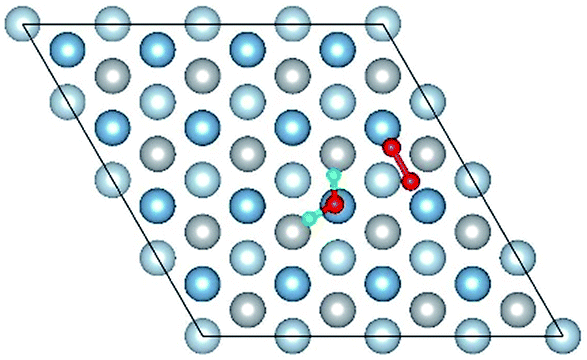
|
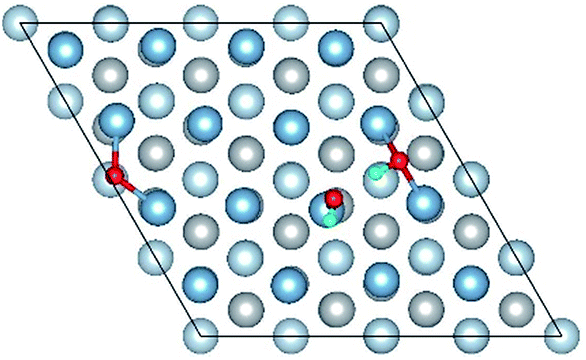
|
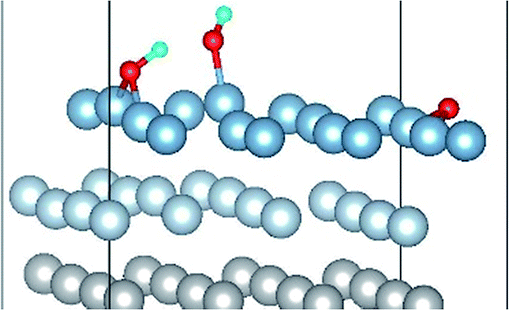
|
| Remark |

|
||
| Initial structure | Final structure | ||
|---|---|---|---|
| Top view | Top view | Side view | |
| Co-4 |
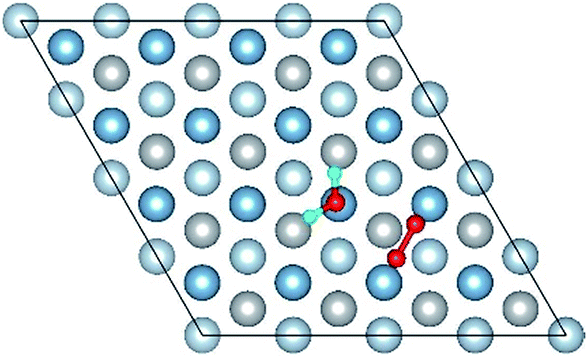
|
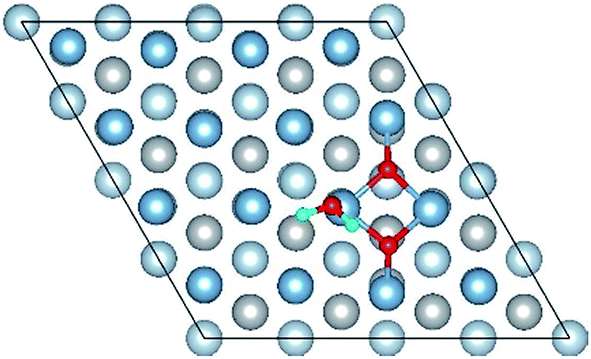
|
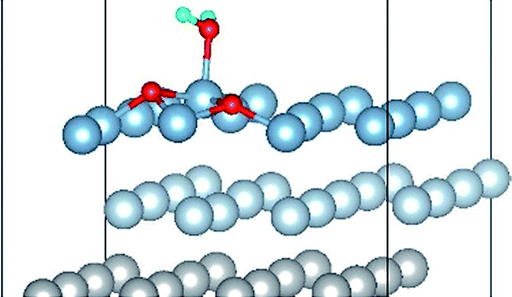
|
| Co-5 |
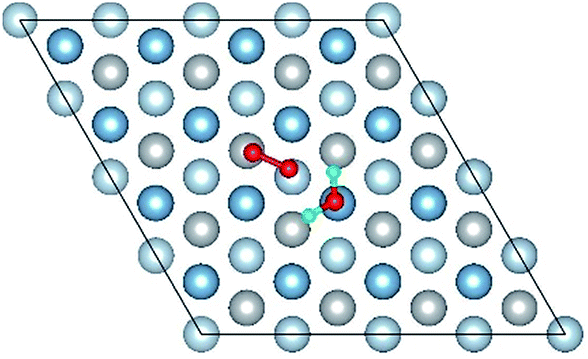
|
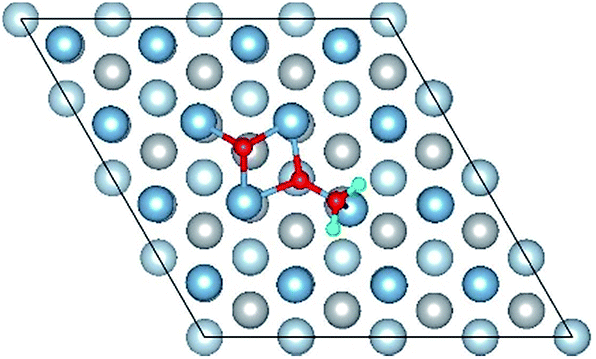
|
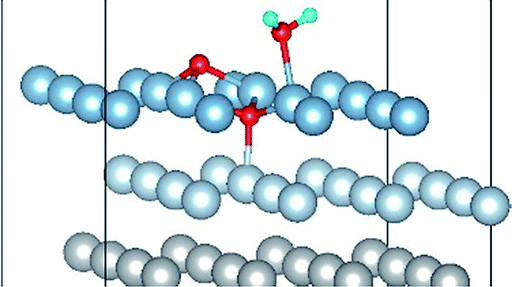
|
| Co-6 |
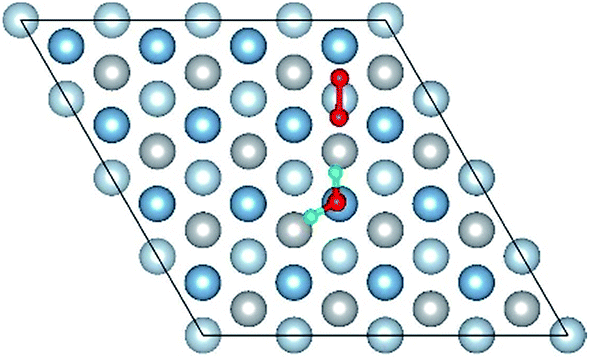
|
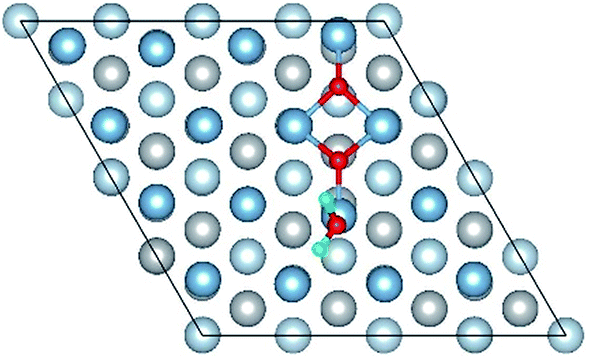
|
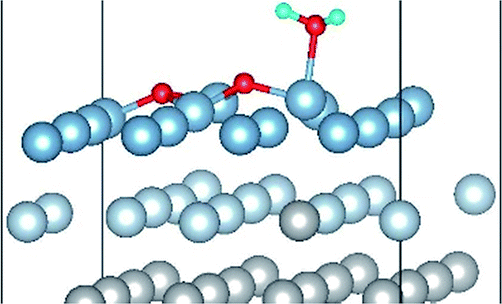
|
| Co-7 |
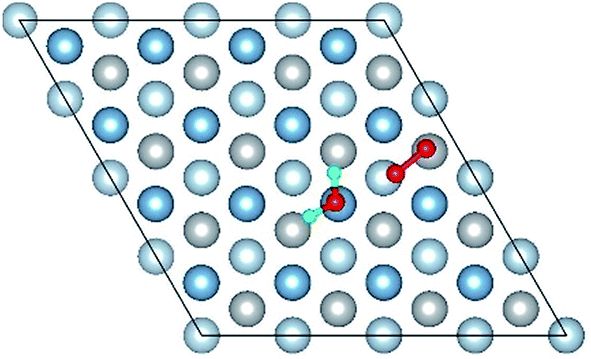
|
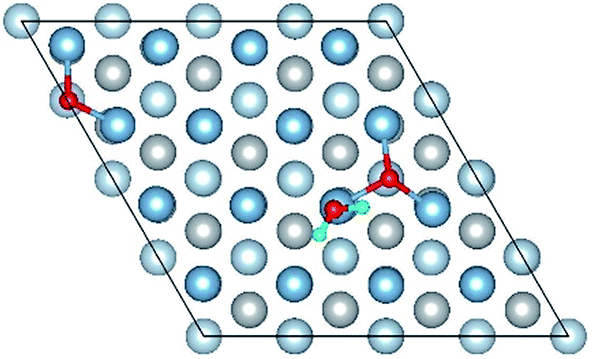
|
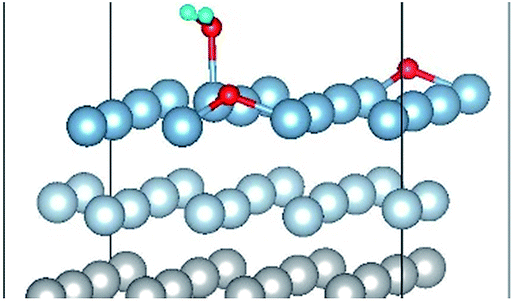
|
| Co-8 |
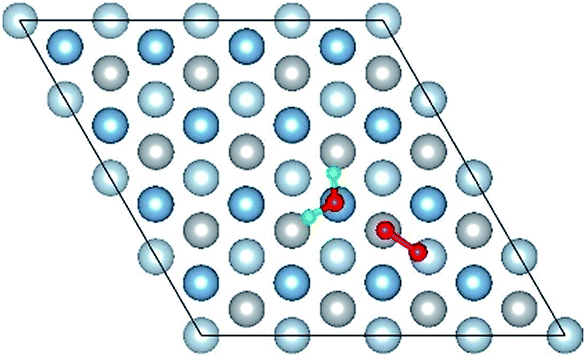
|
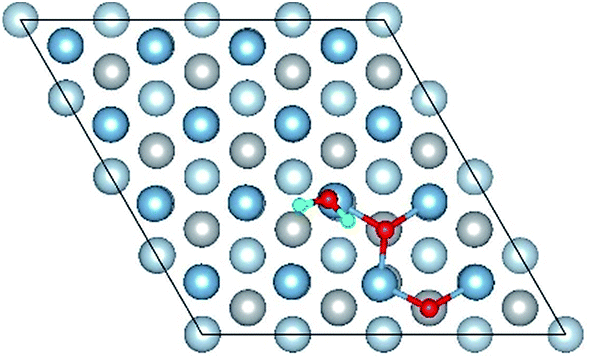
|
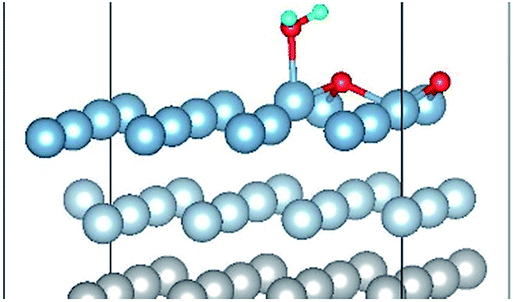
|
| Remark |

|
||
| Adsorbed structure | Distance/Å | Angle ∠HOH/° | E ads/eV | Charge transfer | |||
|---|---|---|---|---|---|---|---|
| Al–O(H2O/OH) | Al–O(O2) | H–O(H2O/OH) | Surface + O2 → H2O | ||||
| Top | Bridge | ||||||
| Co-1 | 1.81 | 1.88 | 1.84 | 1.00 | — | 9.01 | 0.154 |
| Co-2 | 1.81 | 1.86 | 1.84 | 1.01 | — | 9.33 | 0.133 |
| Co-3 | 1.81 | 1.88 | 1.84 | 1.00 | — | 9.00 | 0.163 |
| Co-4 | 2.01 | — | 1.85 | 0.98 | 108.1 | 9.32 | 0.026 |
| Co-5 | 2.07 | — | 1.88 | 0.98 | 108.5 | 9.12 | 0.085 |
| Co-6 | 2.08 | — | 1.85 | 0.98 | 108.5 | 8.95 | 0.037 |
| Co-7 | 2.07 | — | 1.86 | 0.98 | 109.9 | 9.25 | 0.045 |
| Co-8 | 2.07 | — | 1.86 | 0.98 | 108.6 | 9.78 | 0.040 |
The co-adsorbed energy was calculated referenced by O2 and H2O molecule according following equation:
| Eco−ad = Eo2 + EH2O + EAl(111) − Eco−ads/sub | (2) |
E o2, EH2O and EAl(111) are the total energy of the isolated O2 molecule, H2O molecule and the relaxed clean slab, and Eco−ads/sub is the total energy of the surface covered with the adsorbed O2 and H2O. The co-adsorption energy of H2O and O2 on clean Al(111) surface contains three parts: (i) the dissociated adsorption energy of O2, (ii) the molecular or dissociated adsorption energy of H2O and (iii) the interaction energy of O2 and H2O during the reaction process. According to the co-adsorbed structure of the dissociation O2 and H2O (Co-1, Co-2 and Co-3), the adsorption energies of OH on top and bridge site are calculated, which are 1.21 eV and 1.19 eV. The dissociated adsorption energy of O2 is 8.43 eV when the two O atoms adsorbed on the nearest neighbor fcc sites and Eads = 0.40 eV for one H2O adsorption. Compared with the co-adsorption energies of H2O and O2 (Table 5) and the adsorption energy of single O2 on Al(111) surface, the co-adsorption energies are basically contributed by the O2 adsorption. That is why the co-adsorption energy is similar for the dissociated and molecular adsorption of H2O. Similarly, most of charges of surface transfer to O2 molecule to make it dissociated. Fig. 5 shows the difference charge density of co-adsorbed structures (Co-1 and Co-5). Charges transfer from Al atoms and H atoms to O atoms. The charge transfer from surface and O2 to H2O of Co-1 is obviously larger than that of Co-5 indicating that offering enough charge to H2O is one of the necessary condition for the dissociation. Otherwise, H2O will remain the molecular form on surface.
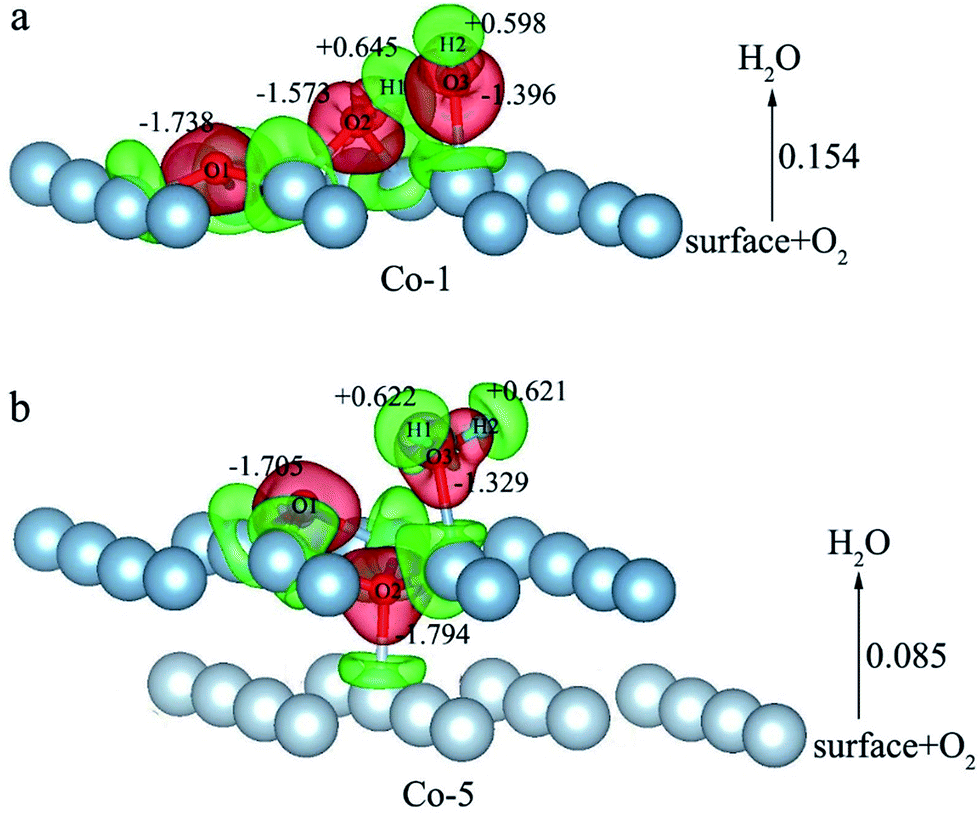 | ||
| Fig. 5 The difference charge density of structures of Co-1 and Co-5 for the co-adsorption of H2O and O2 on Al(111) surface. | ||
4. Conclusions
First-principles calculations based on vdW-DFT have been perform to investigate the co-adsorption behavior of H2O and O2 molecule on Al(111) surfaces. Two types models, adsorption structures of H2O on O(fcc) pre-adsorbed Al(111) surface and co-adsorbed structures of H2O and O2 on clean Al(111) surface, are selected and their interaction carefully analyzed. The key results of our study are:1. O atoms can stable adsorb on fcc sites of Al(111) surface and their adsorption energy referenced single O atom increases with the increasing O coverage. The adsorbed O atoms change the electron density of surface which makes the surface work function increases from 0 to 1 ML.
2. As similar as the clean Al(111), the H2O adsorbs preferentially at the top site of surface. The adsorption is strengthened by increasing the adsorbed O coverage on fcc sites. Although the adsorbed O atoms on fcc sites promote the deformation of H2O and strengthened with the increasing O coverage, but they cannot make it dissociated into OH and H.
3. For the co-adsorption of H2O and O2 on clean Al(111) surface, H2O can dissociate into OH and H in Co-1, Co-2 and Co-3 structure and adsorb on surface by molecular form in remaining structures, which indicates that the existence of O2 can make H2O dissociated. A necessary condition dissociation is offering enough charge to H2O molecule, which is larger than 0.1.
To sum up, O2 can dissociate into O atoms and adsorb on surface, while the dissociation adsorption of H2O relates to many factors. The effect of adsorbed O and O2 molecule on the dissociation of H2O is discussed in this study. The reaction mechanism of H2O dissociation concerning other factors will further study in our subsequent work.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 51222106 and No.51671029), the Fundamental Research Funds for the Central Universities (No. 230201306500002) and National Basic Research Program of China (973 Program) (No. 2014CB643300).References
- L. Österlund, I. Zoric-Acute and B. Kasemo, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 15452–15455 CrossRef.
- I. Popova, V. Zhukov and J. T. Yates Jr, Appl. Phys. Lett., 1999, 75, 3108–3110 CrossRef CAS.
- V. Zhukov, I. Popova and J. T. Yates Jr, J. Vac. Sci. Technol., A, 1999, 17, 1727–1732 CAS.
- J. Trost, H. Brune, J. Wintterlin, R. J. Behm and G. Ertl, J. Chem. Phys., 1998, 108, 1740–1747 CrossRef CAS.
- C. Carbogno, J. Behler, A. Groß and K. Reuter, Phys. Rev. Lett., 2008, 101, 096104 CrossRef PubMed.
- J. Behler, B. Delley, S. Lorenz, K. Reuter and M. Scheffler, Phys. Rev. Lett., 2005, 94, 036104 CrossRef PubMed.
- H. Brune, J. Wintterlin, J. Trost, G. Ertl, J. Wiechers and R. J. Behm, J. Chem. Phys., 1993, 99, 2128–2148 CrossRef CAS.
- Y. Yourdshahyan, B. Razaznejad and B. I. Lundqvist, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 075416 CrossRef.
- K. Honkala and K. Laasonen, Phys. Rev. Lett., 2000, 84, 705–708 CrossRef CAS PubMed.
- B. C. Mitrovic and D. J. O'Connor, Surf. Sci., 1998, 405, 261–270 CrossRef CAS.
- H. A. Al-Abadleh and V. H. Grassian, Langmuir, 2003, 19, 341–347 CrossRef CAS.
- J. Paul and F. M. Hoffmann, J. Phys. Chem., 1986, 90, 5321–5324 CrossRef CAS.
- J. E. Crowell, J. G. Chen, D. M. Hercules and J. T. Yates Jr, J. Chem. Phys., 1987, 86, 5804–5815 CrossRef CAS.
- F. Y. Guo, C. G. Long, J. Zhang, Z. Zhang, C. H. Liu and K. Yu, Appl. Surf. Sci., 2015, 324, 584–589 CrossRef CAS.
- F. P. Netzer and T. E. Madey, Surf. Sci., 1983, 127, 102–109 CrossRef.
- E. M. Stuve, R. J. Madix and C. R. Brundle, Surf. Sci., 1984, 146, 155–178 CrossRef CAS.
- W. T. Wallace and R. L. Whetten, J. Am. Chem. Soc., 2002, 124, 7499–7505 CrossRef CAS PubMed.
- G. Ramis, G. Busca, F. Bregani and P. Forzatti, Appl. Catal., 1990, 64, 259–278 CrossRef CAS.
- J. E. Crowell, E. L. Garfunkel and G. A. Somorjai, Surf. Sci., 1982, 121, 303–320 CrossRef CAS.
- G. L. Griffin and J. T. Yates Jr, J. Chem. Phys., 1982, 77, 3751–3758 CrossRef CAS.
- A. A. Koverga, S. Frank and M. T. M. Koper, Electrochim. Acta, 2013, 101, 244–253 CrossRef CAS.
- X. Wei, C. F. Dong, Z. H. Chen, K. Xiao and X. G. Li, Appl. Surf. Sci., 2015, 355, 429–435 CrossRef CAS.
- L. A. S. RIes, M. D. C. Belo, M. G. S. Ferreira and I. L. Muller, Corros. Sci., 2008, 50, 676–686 CrossRef CAS.
- X. Wei, C. Dong, Z. Chen, K. Xiao and X. Li, RSC Adv., 2016, 6, 56303–56312 RSC.
- G. Roman-Perez and J. M. Soler, Phys. Rev. Lett., 2009, 103, 096102 CrossRef PubMed.
- G. Kresse and J. Hafner, Phys. Rev. Lett., 1993, 47, 558–561 CAS.
- G. Kresse and J. Furthmüller, J. Phys. Chem. B, 1996, 54, 11169–11186 CAS.
- W. Kohn and L. J. Sham, Phys. Rev. A, 1965, 140, 1133–1138 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. Klimeš, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131 CrossRef.
- J. Klimeš, D. R. Bowler and A. Michaelides, J. Phys.: Condens. Matter, 2010, 22, 022201 CrossRef PubMed.
- C. Gattinoni and A. Michaelides, Faraday Discuss., 2015, 180, 439–458 RSC.
- S. Gautier, S. N. Steinmann, C. Michel, C. Michel, P. Fleurat-Lessard and P. Sautet, Phys. Chem. Chem. Phys., 2015, 17, 28921–28930 RSC.
- O. I. Malyi, P. Thiyam, M. Boström, M. Boström and C. Persson, Phys. Chem. Chem. Phys., 2015, 17, 20125–20133 RSC.
- H. Monkhorst and J. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2008, 41, 653–658 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules-A Quantum Theory, Oxford University Press, New York, 1990 Search PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- X. Lei and L. Yang, Computes and applied chemistry, 2011, 28, 793–796 CAS.
- D. Costa, T. Ribeiro and F. Mercuri, Adv. Mater. Interfaces, 2014, 1, 1300072 CrossRef.
- A. Kiejna and B. I. Lundqvist, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 085405 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |