
All-solid-state nanocomposite electrolytes composed of an ionic polymer with polar groups and surface-modified SiO2 nanoparticles for dye-sensitized solar cells†
Qizhang Huangab,
Jifu Shi*a,
Xueqing Xua,
Leilei Wanga,
Liuwen Zhonga,
Yaoming Suna,
Hai Wanga,
Gang Xua and
Yanqing Ge*c
aGuangzhou Institute of Energy Conversion, Key Laboratory of Renewable Energy and Gas Hydrate, Guangdong Key Laboratory of New and Renewable Energy Research and Development, Chinese Academy of Sciences, Guangzhou 510640, P. R. China. E-mail: shijf@ms.giec.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cSchool of Chemical Engineering, Taishan Medical University, Taian 271000, China. E-mail: yanqingge@aliyun.com
First published on 17th August 2016
Abstract
Solid-state electrolytes based on ionic polymer (IP)–SiO2 nanocomposite are prepared for dye-sensitized solar cells (DSCs). An IP with a cationic main chain and polar ester and carbonyl groups is synthesized from 2,3-dichloropropionic acid methyl ester and N,N′-carbonyldiimidazole. SiO2 nanoparticles are chemically modified by silane coupling agents KH550 and KH570. The polar groups in the IP and on the surface of the modified SiO2 can facilitate the ionization of iodide in the electrolyte and increase the number of charge carriers. Moreover, according to the principle of ‘like dissolves like’, SiO2 modified by polar groups can be compatible with the IP and yield a long-term stability for the electrolyte. Eventually, the solid-state electrolyte containing IP and modified SiO2 achieves a high conductivity of 3.05 mS cm−1. The all-solid-state DSCs with the composite electrolyte have favorable stability and photo-to-current conversion efficiencies of 5.03% at 30 mW cm−2 and 4.86% at 100 mW cm−2.
1. Introduction
Dye-sensitized solar cells (DSCs) have attracted a great deal of attention as photovoltaics due to their outstanding advantages of low-cost, easy fabrication, and relatively high efficiency.1,2 The electrolyte, acting as a charge transporter, has significant impact on both the power conversion efficiency and long-term stability of DSCs.3 So far, although the overall conversion efficiencies of DSCs based on traditional liquid-state electrolytes have reached 13%,4 these liquid electrolytes suffer from liquid leakage problems and evaporation of the liquid solvent,5 which lead to less-than-satisfactory stability and limitations for their outdoor application. On the other hand, gelation of organic solvent-based or ionic liquid-based liquid electrolytes with polymers, nanoparticles, or low molecular mass organogelators can produce quasi-solid-state electrolytes, which can mitigate the instability of DSCs to some extent.6–8 However, evaporation of solvent and leakage of liquid component are still inevitable problems in quasi-solid-state electrolytes. In order to find out a perfect solution, solid-state electrolytes without liquid or volatile component have been developed, such as p-type inorganic semiconductors (CuI, CuSCN, CsSnI3, etc.),9–11 organic hole-transport materials (P3HT, Spiro-OMeTAD, PEDOT, etc.),12–15 and ionic polymer-based electrolytes.16 Among them, ionic polymer-based electrolytes combine the high conductivity of ionic liquids and favorable mechanical stability of solid polymers. Thus, ionic polymer-based electrolytes exhibit a picture of the prospective industrial application in the DSCs.On this basis, we have proposed alkyloxy-imidazolium iodide-based ionic polymer (AIIP) electrolyte and proved that the introduction of SiO2 nanoparticles in ionic polymer electrolyte could lead to an increase in conductivity from 0.107 to 0.151 mS cm−1.17 The efficiency of DSCs with AIIP/SiO2 nanocomposite electrolyte attains 4.12%.17 Nevertheless, we found that the conductivity of our AIIP/SiO2 nanocomposite electrolyte would decay by 50% after aging for 1000 h due to the aggregation of the SiO2 nanoparticles. At the same time, the stability of DSCs is closely related to the stability of the electrolyte. Therefore, it is necessary to prepare a kind of solid electrolyte with high conductivity and preferable stability for high-performance all-solid-state DSCs. To date, it is still a challenge to prepare solid electrolytes for all-solid-state DSCs with high η and considerable stability.18–20
In this paper, an ionic polymer (IP) is synthesized through 2,3-dichloropropionic acid methyl ester with ester group (–COOCH3) and N,N′-carbonyldiimidazole with carbonyl group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), where ester and carbonyl groups possess high dielectric constant. The synthesized IP with polar groups can facilitate the ionization of iodide (like 1,2-dimethyl-3-propylimidazolium iodide) in the electrolyte, which consequently increases the number of charge carriers and improves the conductivity. Meanwhile, nano-SiO2 particles are chemically modified by silane coupling agents KH550 and KH570 to introduce polar amino (–NH2) and ester (–COO–) groups, respectively. These modified nano-SiO2 particles with polar groups promote the ionization of iodide as well. In addition, according to the principle of ‘like dissolves like’, the modified nano-SiO2 nanoparticles can mix with IP compatibly and stably thanks to the polarity of the polar groups similar to those in the IP. Eventually, all-solid-state DSCs with the IP electrolyte yield a high overall efficiency and satisfactory long-term stability.
O), where ester and carbonyl groups possess high dielectric constant. The synthesized IP with polar groups can facilitate the ionization of iodide (like 1,2-dimethyl-3-propylimidazolium iodide) in the electrolyte, which consequently increases the number of charge carriers and improves the conductivity. Meanwhile, nano-SiO2 particles are chemically modified by silane coupling agents KH550 and KH570 to introduce polar amino (–NH2) and ester (–COO–) groups, respectively. These modified nano-SiO2 particles with polar groups promote the ionization of iodide as well. In addition, according to the principle of ‘like dissolves like’, the modified nano-SiO2 nanoparticles can mix with IP compatibly and stably thanks to the polarity of the polar groups similar to those in the IP. Eventually, all-solid-state DSCs with the IP electrolyte yield a high overall efficiency and satisfactory long-term stability.
2. Experimental
2.1. Materials
All experimental reagents were of analytical grade. 2,3-Dichloropropionic acid methyl ester (Aladdin) and N,N′-carbonyldiimidazole (Aladdin) were used as received. SiO2 nanoparticles with particle size of 7–10 and 20–30 nm for surface modification were supplied by Degussa AG of Germany (see Fig. S1, ESI,† for TEM images of the SiO2 nanoparticles). TiO2 powders (P25, Degussa AG of Germany) consisted of 30 wt% rutile and 70 wt% anatase. The conductive glass of fluorine-doped SnO2 (FTO, 14 Ω sq−1) was purchased from Asahi Glass. Silane coupling agents (KH550 and KH570) were supplied by Nanjin Jing Tian Wei Chemical Co., Ltd. Di-tetrabutylammoniumcis-bis(isothiocyanato)bis(2,2′-bipyridyl-4,4′-dicarboxylato)ruthenium(II) (N719) was purchased from Dyesol Ltd. Other reagents were supplied by Sinopharm Chemical Reagent Co., Ltd.2.2. Preparation of modified nano-SiO2
The preparation process of modified nano-SiO2 is shown in Fig. 1a. SiO2 nanoparticles (in size of 7–10 or 20–30 nm) were degassed under vacuum at 120 °C for 3 h in order to activate its surface chemical bonds. Then, nano-SiO2 and silane coupling agent (KH550 or KH570) at mass ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 were dispersed by toluene and refluxed at 110 °C for 2 h. At last, after washing by alcohol and centrifugation, modified SiO2 was dried under vacuum at 80 °C for 2 h.
:3 were dispersed by toluene and refluxed at 110 °C for 2 h. At last, after washing by alcohol and centrifugation, modified SiO2 was dried under vacuum at 80 °C for 2 h.
 | ||
| Fig. 1 (a) Modification of SiO2 by KH550 and KH570, (b) preparation of all-solid-state electrolyte, and (c) polymerization of 2,3-dichloropropionic acid methyl ester and N,N′-carbonyldiimidazole. | ||
2.3. Preparation of all-solid-state electrolytes
As illustrated in Fig. 1b, 2,3-dichloropropionic acid methyl ester, N,N′-carbonyldiimidazole, 1,2-dimethyl-3-propylimidazolium iodide (DMPII), I2, and modified nano-SiO2 in different proportions (see the mole ratios of DMPII and I2 to the amount of substance of –COOCH3 on 2,3-dichloropropionic acid methyl ester and the mass fraction of modified SiO2 against the electrolyte, in Section 3.2) were mixed in acetone by stirring. Afterwards, the mixtures were refluxed at 50 °C for 2.5 h. During the procedure of reflux, the ionic polymer (IP) was formed by the polymerization reaction between 2,3-dichloropropionic acid methyl ester and N,N′-carbonyldiimidazole with molar ratio of 1:1 (Fig. 1c). Due to the premixing of the starting materials, the IP electrolyte can be expected to be homogeneous so that the iodide and modified SiO2 evenly distribute within the IP. Finally, the all-solid-state electrolyte was obtained after drying under vacuum at 60 °C for eliminating the residue acetone.
2.4. Fabrication of the DSCs
The about 8 μm-thick nanocrystalline TiO2 film was fabricated on a conducting glass substrate using P25 (medium diameter: 25 nm) by a screen printing technique as the ref. 17. The thickness of the nanocrystalline TiO2 films was controlled by four repetition of screen-printing one time (about 2 μm), which has been measured by Dektak-150 surface profiler in our previous work.17 The TiO2 film with 0.16 cm2 (4 mm × 4 mm) area was sintered at 450 °C for 30 min. After cooling down to 80 °C, it was immersed in an acetonitrile solution containing 5 × 10−4 M of ruthenium dye (N719) for overnight to adsorb dye molecules. Platinized FTO counter electrode was prepared by spin coating 0.05 M chloroplatinic acid in alcohol and sintering at 390 °C for 30 min. The cell was fabricated by clamping a slice of the solid electrolyte between a dye-adsorbed TiO2 photoanode and a platinized FTO counter electrode. Then, the cell was heated at 50 °C for 15 min for better interfacial contact between electrodes and electrolyte.2.5. Characterizations
Infrared absorption spectra were recorded on a Fourier transform infrared (FT-IR) spectrometer (Thermo Fisher Scientific: Nicolet 6700). Thermogravimetric analysis (TGA) was carried out by employing simultaneous thermal analyzer (NETZSCH: STA409C/PC). The Raman spectra were collected by using (HORIBA JY: LabRAM HR800). Power conversion efficiency of DSC with 0.16 cm2 active area was evaluated using a 3A-classsolar simulator (ABET: Sun 3000) which was corrected by standard silicon solar cell before test. The photocurrent–voltage characteristics of the DSC were obtained using a Keithley 2420 digital source meter (Keithley, USA). Electrochemical impedance spectroscopy (EIS) measurements were recorded with electrochemical workstation (Autolab PGSTAT302N). The cross section morphologies of TiO2 layers were observed using scanning electronic microscope (HITACHIS-4800 FESEM). The conductivity of the IP electrolyte was determined by impedance measurement (electrochemical workstation of Autolab PGSTAT302N), which is described as follows. The electrolyte was sandwiched between two mirror-finished stainless steel electrodes using a Teflon ring spacer in a constant volume cylindrical cell. The conductivity was calculated from the ionic resistance (R), which was obtained from the intercept on the real part of the impedance in the complex plane, by the following equation σ = Kcell/R. Kcell is the cell constant tested by using standard potassium chloride solution.3. Results and discussion
3.1. Characterization of ionic polymer and modified nano-SiO2
The FT-IR spectra are used to investigate the structure of the IP and modified nano-SiO2. There are two C–Cl bonds in the molecular structure of 2,3-dichloropropionic acid methyl ester. In Fig. S2,† the characteristic peak of C–Cl bond in 2,3-dichloropropionic acid methyl ester can be seen at 704 cm−1.21 Through the reaction between 2,3-dichloropropionic acid methyl ester and N,N′-carbonyldiimidazole, the covalent C–Cl bonds of 2,3-dichloropropionic acid methyl ester are transformed into ionic bonds (as shown in Fig. 1c). In the FTIR spectra of the synthesized IP, no peak can be observed at around 704 cm−1, indicating polymerization of 2,3-dichloropropionic acid methyl ester and N,N′-carbonyldiimidazole. In addition, in order to further prove the existence of chloride ions in IP, we added drops of AgNO3 aqueous solution into the obtained IP and found white precipitate (AgCl).Fig. 2a shows the infrared spectra of SiO2 particles in size of 7–10 nm, which are naked SiO2 powder (noted as naked SiO2), KH550-modified SiO2 (noted as a-KH550), and KH570-modified SiO2 (noted as a-KH570), respectively. The peaks at ∼1103, ∼808, and ∼470 cm−1 are ascribed to the stretching vibration of Si–OH, the asymmetric stretching vibration of Si–O–Si, and the symmetric stretching and bending vibrations of Si–O–Si, respectively. The broad absorption peak at ∼3442 cm−1 is attributed to Si–OH and the adsorbed H2O. The band at around 1635 cm−1 corresponds to –OH bending vibration of the adsorbed H2O. The peak at around 954 cm−1 is the out-of-plane bending vibration of –OH.22 These results indicate that there are adsorbed water and hydroxy groups (–OH) in the three SiO2 samples. Hydroxy groups (–OH) on nano-SiO2 surface provide silane coupling agent with a possibility for chemical modification. The possible mechanism for the chemical modification is the dehydration between the hydroxy groups (–OH) on nano-SiO2 surface and in the hydrolysed silane coupling agent (KH550 or KH570).22
 | ||
| Fig. 2 FTIR spectra of SiO2 particles in the wavelength of (a) 4000 to 400 cm−1 and (b) 2000 to 400 cm−1 (a-KH550: KH550-modified SiO2 in size of 7–10 nm, a-KH570: KH570-modified SiO2 in size of 7–10 nm), and (c) Raman spectra of the ionic polymer electrolytes with different addition of iodine. | ||
Fig. 2b is the amplification of Fig. 2a in the wavenumber range from 2000 cm−1 to 400 cm−1. For a-KH550, the peak at 1569 cm−1 is ascribed to N–H bond, which indicates that KH550 is successfully grafted to nano-SiO2.23 For a-KH570, the absorption peak at 1703 cm−1 is attributed to CO bond, which verifies that the surface of nano-SiO2 is modified by KH570.22 Thermogravimetric analysis is used to evaluate the adsorption amount of silane coupling agent on the surface of nano-SiO2. The TGA curves of modified SiO2 particles are displayed in Fig. S3.† All the modified SiO2 samples obviously lose weight at around 400 °C. The weight loss from 200 to 800 °C of a-KH570 (9.08 wt%) is greater than that of a-KH550 (8.05 wt%) due to larger relative molecular mass of KH570 (248.35) than KH550 (221.37). The weight loss from 200 to 800 °C of KH570-modified nano-SiO2 in size of 20–30 nm (noted as b-KH570) is 7.14 wt%, denoting that a-KH570 with a larger surface area has more KH570 adsorbed than b-KH570 does.
3.2. Characterization of electrolytes
For low-fluidity iodide electrolytes, charge transport is mainly governed by Grotthus relay-type mechanism, where a net charge transport is achieved without any net transport of mass.16 The net charge transport is affected by (1) the concentration of iodine and (2) the distance between charge transfer centers (donors and acceptors). Moreover, I2 and I− tend to combine to form polyiodide chains (I3−, I5−,…, I2n+1−) owing to outer-shell electrons of I− and the labile-induced dipole of I2.24 The polyiodide chains as well as I− can serve as donors. Therefore, it is necessary to control the concentration of I2 and DMPII in the electrolyte for desirable conductivity.Fig. 2c shows Raman spectra of the IP electrolyte with different mole ratios of I2/–COOCH3 (where –COOCH3 is on the IP and originally on the 2,3-dichloropropionic acid methyl ester before its polymerization) and the mole ratio of DMPII/–COOCH3 fixed at 0.6. When the mole ratio is ≤0.2, no band can be apparently observed from 100 to 200 cm−1. However, when the mole ratio of I2/–COOCH3 increases from 0.4 to 0.8, the bands at 111 and 145 cm−1 appear, which are respectively ascribed to symmetric and asymmetric vibration modes of I3−,17 implying the formation of I3− in the IP electrolyte. As the mole ratio of I2/–COOCH3 ≥ 1.0, another band at 165 cm−1 ascribed to the vibration of I5− is observed in the Raman spectra, indicating the existence of I5− in the IP electrolyte.17
The influence of the mole ratio of the I2/–COOCH3 on the conductivity of the IP electrolyte is shown in Fig. 3a, where the mole ratio of DMPII/–COOCH3 is fixed at 0.6. The increase of ionic conductivity versus the mole ratio of I2/–COOCH3 displays a three-staged dependence. In the first stage (I), the conductivity remains at about 0.02 mS cm−1 with the mole ratio of I2/–COOCH3 less than 0.2, where only I− is in the electrolyte (Fig. 2c). Because the average distance between I− ions (>0.7 nm, calculated from molecular structure of DMPII) is far greater than the radius of I− ion of 0.22 nm,16 electron exchange reaction is hard to occur, thereby resulting in low conductivity. In the second stage (II), the conductivity slowly increases when the mole ratio of I2/–COOCH3 increases from 0.4 to 0.8, where the charge transport becomes easier due to the formation of I3− with larger radius than I− (see Fig. 2c). Interestingly, in the third stage (III), as the ratio is above 1.0, the conductivity increases very quickly. This phenomenon can be explained by the existence of I5− for much faster charge transport (Fig. 2c). However, surplus iodine as an accepter may increase the oxidation ability of the electrolyte and accelerate back reaction.16,25 Therefore, the mole ratio of I2/–COOH is fixed at 0.8, where the conductivity equals to 0.045 mS cm−1.
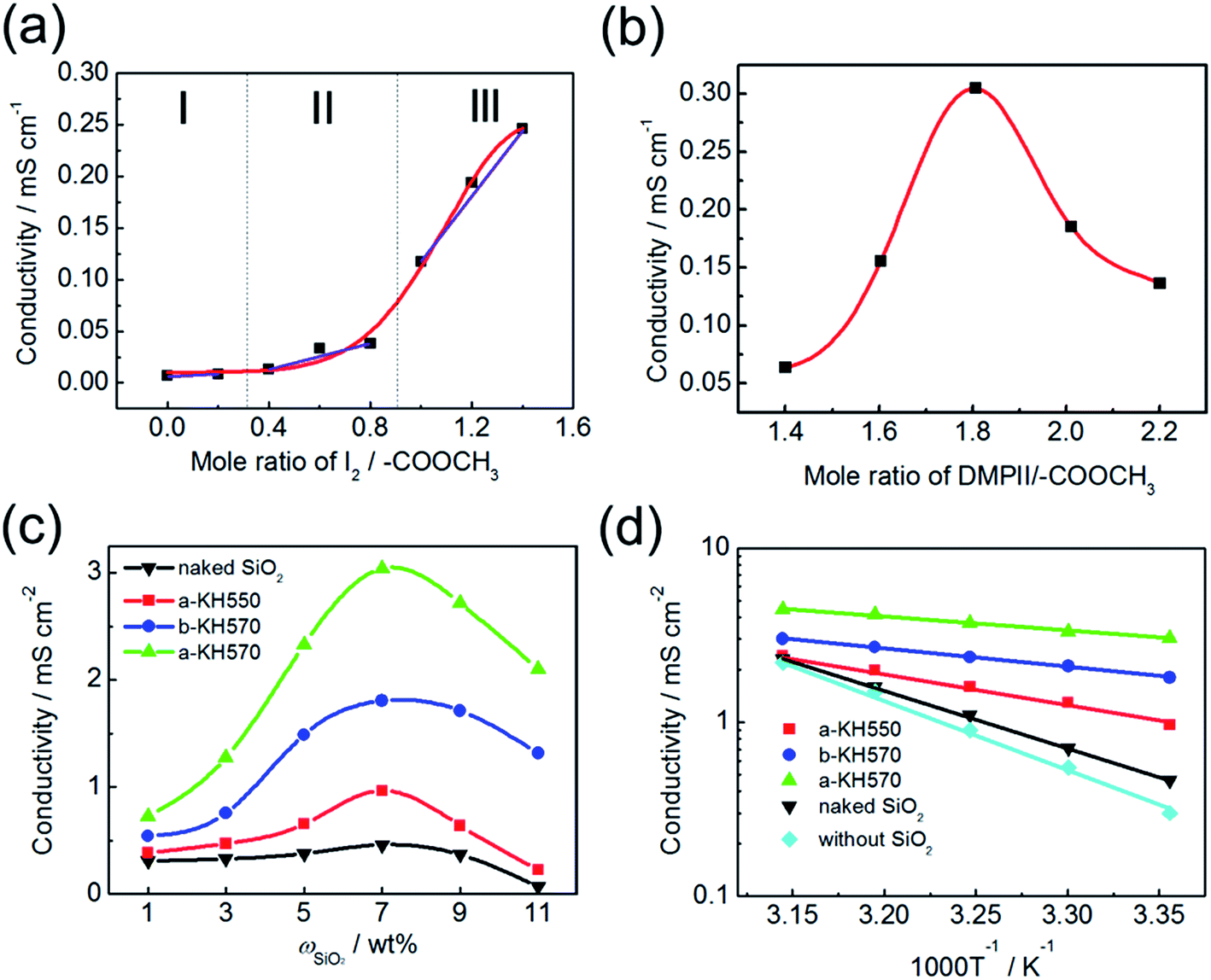 | ||
| Fig. 3 Effects of (a) mole ratio of I2/–COOCH3, (b) mole ratio of DMPII/–COOCH3, (c) weight percentage of SiO2, and (d) temperature on the conductivity of the ionic polymer electrolytes (IP electrolyte with different nano-SiO2 including a-KH550: KH550-modified SiO2 in size of 7–10 nm, a-KH570: KH570-modified SiO2 in size of 7–10 nm, b-KH570: KH570-modified SiO2 in size of 20–30 nm, naked SiO2, and without SiO2). | ||
To the best of our knowledge, the charge transport in the electrolyte is governed by Grotthus relay-type mechanism,16 where charge carriers are located in I2n+1−. Thus, the concentration of I2n+1− has a great influence on conductivity. The addition of DMPII in IP electrolyte can increase the number of charge carriers,17 because the ionization of DMPII can produce more I− for the formation of I2n+1− to endow the IP electrolyte with a better charge transport. Fig. 3b presents the conductivity of the IP electrolyte (with mole ratio of I2/–COOH at 0.8) as a function of the mole ratio of DMPII/–COOCH3. With an increase in the mole ratio of DMPII/–COOCH3, the conductivity gradually increases and then slightly decreases after reaching the maximum conductivity of 0.305 mS cm−1 with the mole ratio of DMPII/–COOCH3 at 1.8. The decrease of the conductivity may result from the aggregates or micro-crystallization of excessive DMPII, blocking electronic transfer.26 The conductivity of the electrolyte based on IP + DMPII (0.305 mS cm−1) is nearly three times as high as that of the previously reported electrolyte based on AIIP + DMPII (0.107 mS cm−1).17 It emphasizes the fact that the introduction of some polar groups (like ester group and carbonyl) into the main chain of the ionic polymer can effectively improve the conductivity of electrolyte.
The effect of modified nano-SiO2 on the conductivity of the electrolyte (at mole ratio of DMPII/I2/–COOCH3 = 1.8/0.8/1) was shown in Fig. 3c. It has been proved that, for the low-fluidity electrolyte containing polyiodide species, the addition of inorganic nanoparticles improves ionic conductivity.27 The conductivity of the IP electrolyte with naked SiO2 increases with the increase of the weight percentage of naked SiO2 from 1 to 7 wt%. However, the conductivity decreases after it reaches the maximum of 0.462 mS cm−1 with 7 wt% of naked SiO2 (Fig. 3c). The increase of the conductivity of the IP electrolyte with naked SiO2 at the beginning for the reason that SiO2 powder can obstruct the contact of the IPs and disrupt their crystallinity, which facilitates the anion transport.28,29 On the other hand, the decrease of the conductivity beyond 7 wt% may be attributed to the aggregation of the excessive SiO2 as an obstacle for charge transport.30 The conductivities of samples with different modified SiO2 follow the same trend as that with naked SiO2 due to the same reason as mentioned above. However, the IP electrolyte with surface modified SiO2 has higher conductivity than that with naked SiO2. The polarity of the additives in electrolyte contributes to the enhancement of salt-dissociation has also been reported.31 As shown in Fig. 4, polar group (–NH2 or –COOR) on the surface of SiO2 as a salt-dissociation enhancer can ionize DMPII by orientation force, through which the negative terminal polar groups attract the cation and repulse the iodine anion to free them out. In other words, the transition of intimate ion-pairs (DMPII) to free ions can offer more charge carriers for conductivity. At the same weight percentage of nano-SiO2, the conductivity of the IP electrolyte with a-KH550 is higher than that with naked SiO2 because polar groups on the surface of a-KH550 are beneficial to ionization of iodide and increase the number of charge carriers. Moreover, the conductivity of IP electrolyte with 7 wt% b-KH570 (1.783 mS cm−1) is increased to a larger degree than that with 7 wt% a-KH550 (0.968 mS cm−1) possibly due to the stronger polarity of KH570 than KH550. Under the condition of the surface modification by KH570, the conductivity of IP electrolyte with 7 wt% of a-KH570 (3.05 mS cm−1) is higher than that with 7 wt% of b-KH570 (1.783 mS cm−1) because smaller particles offer larger surface area for chemical modification, which is in accordance with the larger absorption amount of KH570 from the TGA results.
 | ||
| Fig. 4 Ionization of DMPII by surface modified SiO2 with polar groups. | ||
The temperature dependence of the conductivity of the electrolytes was also measured (Fig. 3d). The conductivity increases with temperature because the electron exchange reaction becomes faster at higher temperatures. The data of temperature dependence of the conductivity can be fitted well by Arrhenius equation:32

The Ea of the IP electrolyte without SiO2 is calculated to be 78.6 kJ mol−1, indicating an inferior ionic conduction. With the addition of naked SiO2 in the IP electrolyte, the Ea is reduced to 63.2 kJ mol−1, because of the construction of electron exchange tunnels by SiO2. With the addition of modified SiO2 a-KH550, b-KH570, and a-KH570 in the electrolyte, the Ea are further decreased to 35.4, 20.1, and 15.3 kJ mol−1, respectively. Some of these values are even comparable to those reported for gel electrolytes (about 20 kJ mol−1).33,34 The lowest Ea of the electrolyte with a-KH570 may be ascribed to the stronger polarity of –COO– and larger modified surface of KH570.
Fig. 5 shows the long-term stability of the IP electrolytes stored at 60 °C for 2000 h. After a stabilizing period, the conductivity of the IP electrolyte without SiO2 decreases to 20% of its initial value approximately after 700 h due to the crystallization of IP. For the IP electrolyte with naked SiO2, the addition of naked SiO2 can hinder the crystallization of IP and alleviate the decrease of the conductivity by the adsorption of the cations on the surface of SiO2.30 However, the conductivity of the IP electrolyte with naked SiO2 can only retain 50% of the origin value after approximately 1200 h because of the aggregation of SiO2. As expected, IP electrolytes with modified SiO2 (a-KH550, b-KH570, and a-KH570) exhibit better long-term stability than those with naked SiO2 and without SiO2. The conductivities of the IP electrolytes (with a-KH550, b-KH570, and a-KH570) almost remain at their original value during the stability test. This suggests that modified SiO2 nanoparticles with polar groups on the surface can disperse homogeneously and have good compatibility with the IP due to the principle of ‘like dissolves like’.
 | ||
| Fig. 5 Long-term stability of different ionic polymer electrolytes. | ||
3.3. Photovoltaic performance and long-term stability of DSCs
Fig. 6a shows photocurrent–voltage (J–V) curves of DSCs with different all-solid-state electrolytes under irradiation at an intensity of 100 mW cm−2. The open-circuit voltages (Voc), short-circuit photocurrent densities (Jsc), fill factors (FF), and the overall energy conversion efficiencies (η) of the DSCs are correspondingly summarized in Table 1. It is notable that the parameters Jsc, Voc, FF, and η of the DSCs based on nano-SiO2 are superior to those of the DSC without SiO2. The magnitude of the Jsc is in the order of a-KH570 (12.89 mA cm−2) > a-KH550 (11.60 mA cm−2) > b-KH570 (4.79 mA cm−2) > naked SiO2 (2.97 mA cm−2) > without SiO2 (1.68 mA cm−2), which is in the same order as the conductivity (Fig. 3d). The difference in the Jsc values mainly arises from variations in the ionic conductivity. | ||
| Fig. 6 Current density–voltage curves of DSCs with different electrolytes at (a) 100 mW cm−2 and (b) 30 mW cm−2, (c) impedance spectra of these devices: Nyquist plots and the equivalent circuits, and (d) long-term stability of the efficiencies of the DSCs. | ||
| Intensity (mW cm−2) | Electrolyte | Jsc (mA cm−2) | Voc (mV) | FF (%) | η (%) |
|---|---|---|---|---|---|
| 100 | a-KH550 | 11.60 | 630 | 63.1 | 4.61 |
| a-KH570 | 12.89 | 608 | 62.0 | 4.86 | |
| b-KH570 | 4.79 | 594 | 61.7 | 1.75 | |
| Naked SiO2 | 2.97 | 555 | 61.0 | 1.01 | |
| Without SiO2 | 1.68 | 553 | 60.0 | 0.56 | |
| 30 | a-KH550 | 4.54 | 536 | 60.7 | 4.92 |
| a-KH570 | 4.83 | 531 | 58.8 | 5.03 | |
| b-KH570 | 1.93 | 525 | 62.5 | 2.11 | |
| Naked SiO2 | 1.61 | 489 | 60.1 | 1.58 | |
| Without SiO2 | 1.19 | 488 | 59.2 | 1.15 |
The magnitude of the Voc is in the order of a-KH550 (630 mV) > a-KH570 (608 mV) > b-KH570 (594 mV) > naked SiO2 (555 mV) > without SiO2 (553 mV). The Voc is determined by the difference between the electron quasi-Fermi level for TiO2 layer and the redox potential of the electrolyte. An upward shift of electron quasi-Fermi level can be realized by minimizing the back reaction at the TiO2/electrolyte interface and hence decreased electron recombination enhances electron accumulation in the TiO2 layer.35 The back reaction is considered to derive from the reduction of I3− by the conduction band electrons of TiO2, which is characterized by the electron recombination resistance at the dyed-TiO2/electrolyte interface (Rct).36 Thus, impedance spectra are employed to study the back reaction at the dyed-TiO2/electrolyte interface. Fig. 6c shows the Nyquist plot of the DSCs measured in the dark under a forward bias of −620 mV, which is close to the value of Voc. The semi-circle in the middle-frequency range corresponds to the recombination process at the dyed-TiO2/electrolyte interface. The equivalent circuit used for fitting the impedance spectra of the DSCs is shown in the inset of Fig. 6c. In the equivalent circuit, the physical meanings of Rs, Zw, Rct, Cμ, Rpt, and CPE1 have been explained in the ref. 37. The increase of Rct can cause the decrease of electron recombination which can increase the electron density of the conduction band of TiO2. Therefore, a higher Voc can be expected by the increase of Rct. The values of Rct which can be obtained by fitting the diameter of the semicircle at the middle-frequency are given as follow: 113 Ω (b-KH570) > 54 Ω (a-KH550) > 45 Ω (a-KH570)> 38 Ω (naked SiO2) > 36 Ω (without SiO2). As shown in Fig. 6c, except for the DSC with b-KH570, the DSCs with modified SiO2 have larger Rct and thus exhibit higher Voc (as listed in Table 1). However, the DSC with b-KH570 has the largest Rct but does not exhibit the highest Voc. This can be ascribed to the poor interfacial contact between the electrolyte and TiO2 due to the larger particle size of b-KH570 (20–30 nm) than the pore size (with average value of 22 nm) of TiO2.38 Fig. 7 shows the scanning electron micrographs of the cross section of the TiO2 layers after introducing IP electrolytes. In Fig. 7a, the porous TiO2 are completely filled with the IP electrolyte without SiO2, which indicates an intimate interfacial contact. Such efficient filling can also be observed for IP electrolytes with naked SiO2, a-KH550, and a-KH570 (in size of 7–10 nm) (Fig. 7b–d). It is noted that SiO2 nanoparticles with diameters from 7 to 10 nm are preferred for a satisfactory contact with TiO2 because they are small enough to penetrate into the pores of the typical nanoporous TiO2 layer whose average pore size is about 22 nm.38 However, in Fig. 7e, porous structure can be still clearly observed within the rough TiO2 layer, suggesting that most of the electrolytes with b-KH570 cannot fill in the porosity, which is the main reason for its largest Rct. The increase of Rct arose from the poor interfacial contact does not contribute to the increase of Voc. Therefore, the Voc of the DSC with b-KH570 is not the highest.
 | ||
| Fig. 7 SEM images of the cross section of the TiO2 layers after introducing different IP electrolytes: (a) without SiO2, (b) with naked SiO2, (c) with a-KH550, (d) with a-KH570, and (e) with b-KH570. | ||
At the intensity of 30 mW cm−2, the magnitude of the Jsc of the DSCs is in the same order of a-KH570 (4.83 mA cm−2) > a-KH550 (4.54 mA cm−2) > b-KH570 (1.93 mA cm−2) > naked SiO2 (1.61 mA cm−2) > without SiO2 (1.19 mA cm−2) as that observed at 100 mW cm−2 (Fig. 6b and Table 1). The Voc of a-KH550, a-KH570, b-KH570, naked SiO2, and without SiO2 are 536 mV, 531 mV, 525 mV, 489 mV, and 488 mV, respectively, again coincide with the trend found for 100 mW cm−2.
In general, the DSC with a-KH570 yields the highest overall η of 4.86% at 100 mW cm−2, which is also higher than the η (4.12%) of the previously reported AIIP-based DSC with SiO2 composites.17 The even higher η of 5.03% is also reached by the DSC with a-KH570 at 30 mW cm−2, which is very close to the reported high efficiencies (5–7%) of all-solid-state DSCs.16,39
The long-term stability of the η of the DSCs (stored at 60 °C for 1000 h) is shown in Fig. 6d. The η of the DSC without SiO2 decreases to 20% of its initial value mainly due to the deterioration of the electrolyte by the crystallization of the IP. However, with the naked SiO2, the η of the DSC shows improved stability and only decreases to 50% of its initial value. As expected, DSCs with modified nano-SiO2 (a-KH550, a-KH570, and b-KH570) show favorable stability of their η due to the principle of ‘like dissolves like’. The η of modified SiO2 do not change much after aging for 1000 h. These results are in accordance with the stability of the σ of the corresponding electrolytes.
4. Conclusions
In summary, we have demonstrated all-solid-state ionic polymer electrolytes consisting of iodine, DMPII, and modified SiO2. Through the optimization of the content of I2, DMPII and modified SiO2 in the electrolyte, the ionic conductivity of 3.05 mS cm−1 was achieved. The polar groups in the ionic polymer and on the surface of modified SiO2 can facilitate the ionization of DMPII and produce more charge carriers. Meanwhile, modified SiO2 can be compatible with the ionic polymer. Consequently, the all-solid-state DSSCs with the IP electrolytes (at the mole ratio of DMPII/I2/–COOCH3 = 1.8/0.8/1 and with 7 wt% KH570-modified SiO2) exhibited remarkably high conversion efficiency of 4.86% at 100 mW cm−2, 5.03% at 30 mW cm−2, and good long-term stability where the efficiency remained nearly the same after aging for 1000 h.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21103194, 51506205), Science and Technology Planning Project of Guangdong Province, China (2014A010106018, 2013A011401011), Guangdong-Hong Kong Joint Innovation Project of Guangdong Province, China (2014B050505015), Special Support Program of Guangdong Province, China (2014TQ01N610), and Solar Photothermal Advanced Materials Engineering Research Center Construction Project of Guangdong Province, China (2014B090904071).References
- L. Song, W. J. Wang, V. Korstgens, D. M. Gonzalez, Y. Yao, N. K. Minar, J. M. Feckl, K. Peters, T. Bein, D. Fattakhova-Rohlfing, G. Santoro, S. V. Roth and P. Muller-Buschbaum, Adv. Funct. Mater., 2016, 26, 1498–1506 CrossRef CAS.
- Q. Z. Huang, J. F. Shi, L. L. Wang, Y. J. Li, L. W. Zhong and G. Xu, Thin Solid Films, 2016, 610, 19–25 CrossRef CAS.
- S. Taya, S. Kuwahara, Q. Shen, T. Toyoda and K. Katayama, RSC Adv., 2014, 4, 21517–21520 RSC.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Gratzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- H. H. Lin, J. D. Peng, V. Suryanarayanan, D. Velayutham and K. C. Ho, J. Power Sources, 2016, 311, 167–174 CrossRef CAS.
- Z. P. Huo, C. N. Zhang, X. Q. Fang, M. L. Cai, S. Y. Dai and K. J. Wang, J. Power Sources, 2010, 195, 4384–4390 CrossRef CAS.
- J. Fan, L. Li, H.-S. Rao, Q.-L. Yang, J. Zhang, H.-Y. Chen, L. Chen, D.-B. Kuang and C.-Y. Su, J. Mater. Chem. A, 2014, 2, 15406–15413 CAS.
- S. N. F. Yusuf, A. D. Azzahari, R. Yahya, S. R. Majid, M. A. Careem and A. K. Arof, RSC Adv., 2016, 6, 27714–27724 RSC.
- E. Edri, H. Cohen and G. Hodes, ACS Appl. Mater. Interfaces, 2013, 5, 5156–5164 CAS.
- B. O'regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- S. Ma, M. W. Shang, L. Y. Yu and L. F. Dong, J. Mater. Chem. A, 2015, 3, 1222–1229 CAS.
- A. Abrusci, R. S. S. Kumar, M. Al-Hashimi, M. Heeney, A. Petrozza and H. J. Snaith, Adv. Funct. Mater., 2011, 21, 2571–2579 CrossRef CAS.
- P. Liu, B. Xu, K. M. Karlsson, J. B. Zhang, N. Vlachopoulos, G. Boschloo, L. C. Sun and L. Kloo, J. Mater. Chem. A, 2015, 3, 4420–4427 CAS.
- J. B. Zhang, L. Yang, Y. Shen, B. W. Park, Y. Hao, E. M. J. Johansson, G. Boschloo, L. Kloo, E. Gabrielsson, L. C. Sun, A. Jarboui, C. Perruchot, M. Jouini, N. Vlachopoulos and A. Hagfeldt, J. Phys. Chem. C, 2014, 118, 16591–16601 CAS.
- J. W. Lee, T. Y. Lee, P. J. Yoo, M. Gratzel, S. Mhaisalkar and N. G. Park, J. Mater. Chem. A, 2014, 2, 9251–9259 CAS.
- J. H. Wu, S. Hao, Z. Lan, J. M. Lin, M. L. Huang, Y. F. Huang, P. J. Li, S. Yin and T. Satot, J. Am. Chem. Soc., 2008, 130, 11568–11572 CrossRef CAS PubMed.
- J. F. Shi, L. Wang, Y. L. Liang, S. J. Peng, F. Y. Cheng and J. Chen, J. Phys. Chem. C, 2010, 114, 6814–6821 CAS.
- L. Y. Chang, C. P. Lee, C. T. Li, M. H. Yeh, K. C. Ho and J. J. Lin, J. Mater. Chem. A, 2014, 2, 20814–20822 CAS.
- S. Venkatesan, S. C. Su, S. C. Kao, H. Teng and Y. L. Lee, J. Power Sources, 2015, 274, 506–511 CrossRef CAS.
- L. Song, W. J. Wang, V. Korstgens, D. M. Gonzalez, Y. Yao, N. K. Minar, J. M. Feckl, K. Peters, T. Bein, D. Fattakhova-Rohlfing, G. Santoro, S. V. Roth and P. Muller-Buschbaum, Adv. Funct. Mater., 2016, 26, 1498–1506 CrossRef CAS.
- M. C. Reina, R. Boese, M. Ge, S. E. Ulic, H. Beckers, H. Willner and C. O. Della Vedova, J. Phys. Chem. A, 2008, 112, 7939–7946 CrossRef CAS PubMed.
- H. B. Wang, E. Y. Chen, X. B. Jia, L. J. Liang and Q. Wang, Appl. Surf. Sci., 2015, 349, 724–732 CrossRef CAS.
- Z. Geng, J. Y. Ba, S. L. Zhang, J. S. Luan, X. Jiang, P. F. Huo and G. B. Wang, J. Mater. Chem., 2012, 22, 23534–23540 RSC.
- P. Deplano, J. R. Ferraro, M. L. Mercuri and E. F. Trogu, Coord. Chem. Rev., 1999, 188, 71–95 CrossRef CAS.
- H. X. Wang, X. Z. Liu, Z. X. Wang, H. Li, D. M. Li, Q. B. Meng and L. Q. Chen, J. Phys. Chem. B, 2006, 110, 5970–5974 CrossRef CAS PubMed.
- G. Shang, J. Wu, S. Tang, M. Huang, Z. Lan, Y. Li, J. Zhao and X. Zhang, J. Mater. Chem., 2012, 22, 25335–25339 RSC.
- K. M. Lee, P. Y. Chen, C. P. Lee and K. C. Ho, J. Power Sources, 2009, 190, 573–577 CrossRef CAS.
- S. Yanagida, C. R. Chim., 2006, 9, 597–604 CrossRef CAS.
- T. Stergiopoulos, I. M. Arabatzis, G. Katsaros and P. Falaras, Nano Lett., 2002, 2, 1259–1261 CrossRef CAS.
- M.-S. Kang, K.-S. Ahn and J.-W. Lee, J. Power Sources, 2008, 180, 896–901 CrossRef CAS.
- T. Mizumo, T. Kajihara, T. Yamada and J. Ohshita, Polym. Adv. Technol., 2013, 24, 705–714 CrossRef CAS.
- J. Wu, S. Hao, Z. Lan, J. Lin, M. Huang, Y. F. Huang, L. Fang, S. Yin and T. Sato, Adv. Funct. Mater., 2007, 17, 2645–2652 CrossRef CAS.
- J. Shi, S. Peng, J. Pei, Y. Liang, F. Cheng and J. Chen, ACS Appl. Mater. Interfaces, 2009, 1, 944–950 CAS.
- H. Choe, B. Carroll, D. Pasquariello and K. Abraham, Chem. Mater., 1997, 9, 369–379 CrossRef CAS.
- M. L. Cai, X. Pan, W. Q. Liu, J. Sheng, X. Q. Fang, C. N. Zhang, Z. P. Huo, H. J. Tian, S. F. Xiao and S. Y. Dai, J. Mater. Chem. A, 2013, 1, 4885–4892 CAS.
- K. Kalyanasundaram and M. Grätzel, Coord. Chem. Rev., 1998, 177, 347–414 CrossRef CAS.
- F. L. Guo, X. P. Liu, Y. Ding, F. T. Kong, W. C. Chen, L. Zhou and S. Y. Dai, RSC Adv., 2016, 6, 13433–13441 RSC.
- L. Hu, S. Dai, J. Weng, S. Xiao, Y. Sui, Y. Huang, S. Chen, F. Kong, X. Pan and L. Liang, J. Phys. Chem. B, 2007, 111, 358–362 CrossRef CAS PubMed.
- H. Wang, X. Zhang, F. Gong, G. Zhou and Z. S. Wang, Adv. Mater., 2012, 24, 121–128 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra12787h |
| This journal is © The Royal Society of Chemistry 2016 |