
Characterization of depolymerized lignin and renewable phenolic compounds from liquefied waste biomass†
Abstract
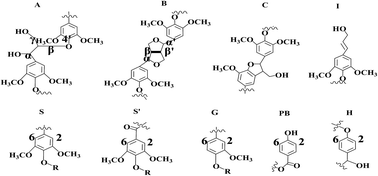
This investigation aimed to analyze the renewable phenolic compounds that separate from liquefied mason pine. One-step thermal conversion of biomass to phenolic products from waste mason pine using an acidic catalyst and methanol was accomplished under mild conditions. Three fractions (fractions 1#, 2#, and 3#) of phenolic compounds with high added-value were extracted with water–organic solvent from liquefied oil via a stepwise fractionation process. The structural features of three phenolic compounds and depolymerized lignin were analyzed and identified by a combination of heteronuclear single quantum correlation-nuclear magnetic resonance, gel permeation chromatography, Fourier transform infrared spectroscopy, and thermogravimetric analysis, showing interesting functionalities for biochemical and biofuel applications. In this investigation, guaiacyl (G) and p-hydroxybenzoate (PB) aromatics were the basic units of three phenolic compound fractions. Etherified syringyl aromatics were evident in fractions 1# and 3#. There were only single-aromatic-ring units (such as G and PB units) in phenolic compound fraction 2#. In the aromatic region, the absence of β-O-4′ ether linkage, resinol, and phenylcoumaran units in three phenolic compound fractions indicated that depolymerization of lignin occurred during the liquefied biomass process. The molecular weights of three phenolic compound fractions were significantly different (797, 249, and 497, respectively) along with fractions 1#, 2#, and 3#. The phenolic compounds could be separated into a high, lower, and lowest molecular weight fraction in this study. As evidenced by GC-MS spectra, the three phenolic compound fraction products and depolymerized lignin were mainly comprised of phenolic derivatives, such as 3-methyl-4-ethylphenol, 4-ethyl-2-methoxyphenol, and 3-methylcatechol. Percentages of the total phenols and derivatives in the three phenolic compound fractions and depolymerized lignin were 77.59%, 81.76%, 80.19%, and 78.86%, respectively. Therefore, the three phenolic compound fractions were clearly quantified and valuable, and can be used as chemical products.
 Please wait while we load your content...
Please wait while we load your content...

