
DNA mediated electrocatalytic enhancement of α-Fe2O3–PEDOT–C-MoS2 hybrid nanostructures for riboflavin detection on screen printed electrode†
C. Sumathia,
P. Muthukumarana,
P. Thivyaa,
J. Wilson*a and
G. Ravib
aPolymer Electronics Lab, Department of Bioelectronics and Biosensors, Alagappa University, Karaikudi-630004, Tamilnadu, India. E-mail: wilson.j2008@yahoo.com
bPhotonic Crystals Lab, Department of Physics, Alagappa University, Karaikudi-630 004, Tamilnadu, India
First published on 12th August 2016
Abstract
A facile synthesis of iron oxide nanorods and PEDOT(poly(3,4-ethylenedioxythiophene)) nanospheres on carbon supported MoS2 (C-MoS2) is reported for riboflavin (RF) sensing. Furthermore, a novel aqueous-based DNA wrapped on an α-Fe2O3–PEDOT–C-MoS2 scaffold shows high electrocatalytic activity compared to that of the α-Fe2O3–PEDOT–C-MoS2 composite in biosensing. α-Fe2O3–PEDOT–C-MoS2/GCE demonstrates the linear response of RF in the concentration range from 800 nM to 700 μM, with a detection limit of 79 nM (S/N = 3σ/b), whereas the α-Fe2O3–PEDOT–C-MoS2–DNA/GCE composite shows a wider range from 300 nM to 1 mM with a comparatively low detection limit of 5 nM. Similarly, α-Fe2O3–PEDOT–C-MoS2–DNA/SPE exhibits a still wider range from 100 nM to 1 mM, with a detection limit of 12 nM. Interestingly, it is also observed that α-Fe2O3–PEDOT–C-MoS2–DNA/GCE reduces the oxidation potential of RF by 30 mV. Thus, the excellent behavior of the proposed biosensor can be attributed to the unique behavior of DNA, which provides a wider detection range and good electrocatalytic behavior towards RF. The fabricated sensor exhibited highly sensitive and selective detection of RF. Real sample analysis was also executed for human urine, milk powder and pharmaceutical drugs without any preliminary treatment.
1. Introduction
Riboflavin (RF), a well-known vitamin, is the primary redox active component of flavin mononucleotide and flavin adenine dinucleotide in blood plasma; it undergoes a wide variety of cellular processes, such as metabolism of fats, ketone bodies, carbohydrates, and proteins.1,2 Therefore, the determination of trace amounts of RF is important in humans and animals. This, in turn, requires superior methods for determining RF and the development of rapid, selective, and highly sensitive techniques. Among the various techniques, electrochemical methods are an attractive option because they are inexpensive, reliable long-term, reproducible and convenient.Conducting polymers have attracted major attention for their applications in a variety of fields, including photovoltaics,3,4 electronics,5,6 sensing,7,8 electroluminescence9,10 and biosensors,11,12 especially due to their optical properties, structural modification, conductivity and comparably low cost. PEDOT, a recently discovered, excellent conducting polymer, has been reported to exhibit enhanced behavior compared to its counterparts.13,14 Recently, our group investigated the PEDOT polymer matrix for the development of ultra-sensitive label free electrochemical DNA sensors. The fabricated sensor was capable of effectively discriminating single and double base mismatch DNA and completely non-complementary DNA.15
For analogous reasons, nanostructured metal oxides are generating great interest, owing to their enhanced electron-transfer kinetics and strong adsorption, which provide suitable microenvironments for the immobilization of biomolecules and result in improved biosensing characteristics.16–19 Maiyalagan et al.20 reported that nanostructured α-Fe2O3 in fact exhibits noble metal behavior and is also found to possess an intrinsic enzyme-mimicking activity similar to that found in natural horseradish peroxidase. Hence, α-Fe2O3 could be utilized for enzyme free biosensing applications. Very recently, we reported that α-Fe2O3 can play the key role in achieving a minimal detection limit due to its catalytic and enhanced conductive behavior in combination with an Au–Pd system.21
Recently, molybdenum disulfide (MoS2), a newly discovered bandgap-adjustable semiconductor composed of S–Mo–S triple layers, has attracted great interest in the fields of electrochemistry, catalysts, sensors, capacitors, and lithium-ion batteries.23–25 MoS2 has a sandwich structure similar to graphene;22 its electrocatalytic activity originates from the sulfur edges of the MoS2 layers, while the inner planes are catalytically inert. W. H. Hu et al.26 reported the highly active and stable electrocatalytic behavior of the MoSx/GO matrix, owing to its good dispersion and conductivity, for the hydrogen evolution reaction. However, the improvement of MoS2 electrocatalyst is still in its infancy due to challenges such as aggregation phenomena and poor conductivity. To date, several strategies have been proposed for the modification of MoS2 composite materials. Among these, carbon supported MoS2 (C-MoS2) is attractive to facilitate electron transfer, resulting in greatly improved conductivity of the composite. W. H. Hu et al.27 reported that the acid treated carbon nanospheres and MoS2 composite exhibited enhanced conductivity and electrocatalytic activity for the hydrogen evolution reaction compared to MoS2.
Moreover, various types of biomolecules, such as amino acids, nucleic acids, proteins, and peptides, have been utilized as templates for the fabrication of nanostructured materials.28–31 Among the various biomolecules, deoxyribonucleic acid (DNA) is an attractive material to develop inorganic nanostructures because it is inexpensive, well-characterized, and shows controllable and compatible behavior. The phosphate groups and sugar molecules of DNA can bind with different metal/polymer cations through electrostatic interactions.32–36
In this paper, mixed electronic/ionic transport in conducting polymer-metal oxide-dichalcogenide biomolecules is used as a platform to design a hybrid nanostructure, due to a host of novel devices that leverage the blend of these carriers to enrich catalytic activity for RF biosensor applications. The work discussed here addresses two aspects: (i) formation of a co-ordination bond between Fe–S of α-Fe2O3 and the monomer EDOT (3,4-ethylenedioxythiophene), which has a lone pair of electrons and may overlap with the vacant d-orbital of Fe; also, carbon enhances the active S edge sites for the electron transfer process; the unpaired spin density of α-Fe2O3 and C-MoS2 will provide more spacing for insertion of other materials in the composite.37 (ii) PEDOT/DNA and α-Fe2O3/DNA composites exhibit enhanced catalytic activity; however, MoS2/DNA composite has less interaction. In order to enhance the conductive nature of MoS2, carbon supported MoS2 is utilized in this work. As DNA can be an effective intercalator and makes the composite less acidic than the PEDOT-PSS (poly(3,4-ethylenedioxythiophene)-polystyrene sulfonate) system, it is therefore expected that DNA doped biocomposite modified electrodes may also possess catalytic activity towards the oxidation of RF (Scheme 1).
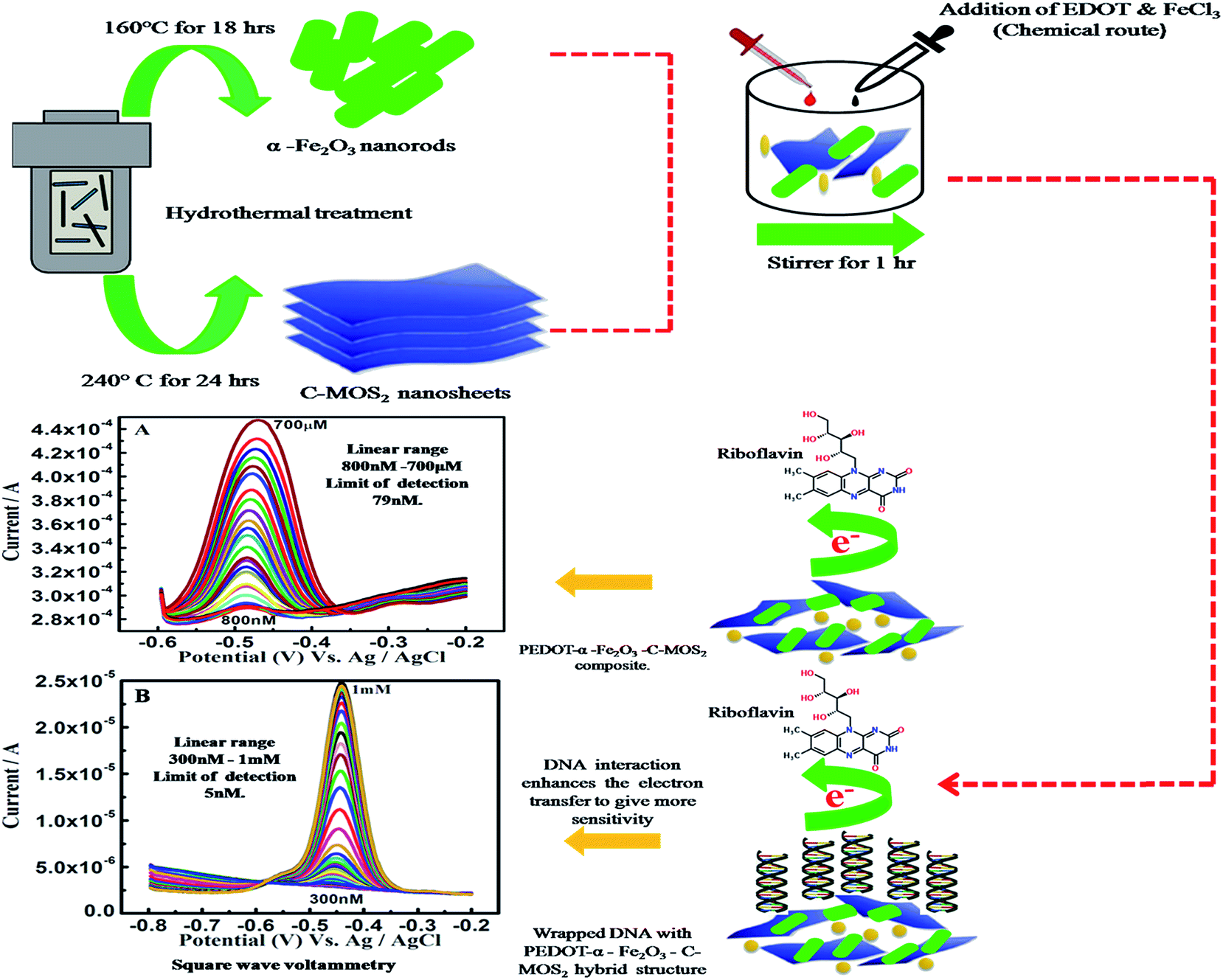 | ||
| Scheme 1 Illustration of preparation of the PEDOT–α-Fe2O3–C-MoS2–DNA hybrid nanostructure and RF detection. | ||
2. Experimental section
2.1 Reagents and materials
Sodium molybdate (Na2MoO4·2H2O) and EDOT monomer were obtained from Sigma Aldrich, India. Glucose, thiourea, RF, and FeCl3 were obtained from Sisco Research Laboratories, Mumbai. Stock 1 mM solution of RF was prepared before use and protected against light. Double-stranded herring testes DNA with an average molecular weight of 50 kbps (base pairs) is simply denoted as DNA throughout this paper. The stock DNA solution (0.06 g per 50 ml) was prepared by mixing a measured amount of DD (doubly distilled) water. Screen printed electrodes (SPE) were from Zensor Technologies, India. The aqueous solutions used throughout were prepared with ultra-pure water obtained from a Millipore system.2.2 Instruments and measurements
The surface morphology of the samples was investigated with a scanning electron microscope (SEM, Carl Zeiss EVO 18); X-ray diffraction (X-RD) was performed using a Bruker Germany D8 Advance instrument with CuKα radiation (1.5418 Å). The Raman spectrum was recorded using an imaging spectrograph (model STR, 500 mm focal length) laser Raman spectrometer (SEKI, Japan). UV studies were performed using a Shimadzu UV-1700 spectrophotometer. Electrochemical experiments were performed using a CHI 6005D electro-chemical workstation (Austin, USA) using a GC working electrode (0.07 cm−2), an Ag/AgCl (3.0 M KCl) reference electrode and a platinum wire auxiliary electrode. All the measurements were carried out in PBS (phosphate buffer solution) as a supporting electrolyte under nitrogen atmosphere at room temperature.2.3 C-MoS2 synthesis
In a typical synthesis, 0.15 g of Na2MoO4·2H2O, 0.50 g of glucose and 0.20 g NH2CSNH2 were dispersed in 30 ml DD water. After stirring for 30 minutes, the reaction solution was transferred into a 50 ml Teflon-lined stainless steel autoclave and heated at 240 °C for 24 h. The autoclave was left to cool naturally. The black precipitate was collected by centrifugation, washed with DD water and ethanol, and dried at 80 °C for 24 h. Afterwards, the as-prepared samples were annealed to 800 °C for 2 h to improve the conductivity of MoS2 under the protection of argon.2.4 α-Fe2O3 nanorods synthesis
Ferric chloride (1.0 g) and sodium hydroxide (NaOH) pellets (0.2 g) were sequentially added to a beaker of 100 ml DD water. Then, this solution was stirred and heated in the autoclave to 160 °C for 18 h. Finally, the heated mixture was centrifuged, washed and dried as earlier.2.5 DNA stock solution preparation
In a typical synthesis, 10 ml of stock DNA solution (0.06 g per 50 ml) was prepared by mixing a measured amount of DD water. The solution containing DNA was stirred using a magnetic stirrer overnight, which is useful to obtain homogeneous DNA solution without any pop-up of adenine and guanine bases in the DNA.2.6 PEDOT–α-Fe2O3–C-MoS2–DNA hybrid nanostructure
200 μl EDOT monomer was dispersed in 50 ml distilled water and stirred for 30 minutes. Next, the oxidizing agent FeCl3 was added to the above solution. Then, C-MOS2 (0.05 g) and α-Fe2O3 (0.05 g) were also mixed into the above solution, which was stirred for 1 h. Finally, 2 ml of DNA from the stock solution was added and heated to 60 °C for 45 min. Then, the sample was used for various electrochemical studies.3. Results and discussion
3.1 Structural analysis
 | ||
| Fig. 1 (A) Raman spectra, (B) XRD patterns of the α-Fe2O3–PEDOT–C-MoS2 composite. (C) UV patterns of the α-Fe2O3–PEDOT–C-MoS2–DNA hybrid nanostructure. | ||
3.2 Morphology analysis
The SEM image in Fig. 2A reveals the well dispersed regular and desirable rod shapes of Fe2O3 with crystalline morphology; the average particle size remains 96.5 nm. Fig. 2B shows the C-MoS2 nanosheets with slightly folded dentations on the edges which considerably enhance the exposure of the active edge sites.47 This is due to the presence of carbon, and it is used to decentralize the MoS2 nanosheets. Fig. 2C depicts the nanospheres of PEDOT; their size is quite uniform (∼36.5 nm). The presence of α-Fe2O3 and PEDOT embedded on the C-MoS2 sheets is seen in Fig. 2D. The DNA wrapped on the α-Fe2O3–PEDOT–C-MoS2 composite is exhibited in Fig. 2E. The size enhancement after coating the α-Fe2O3–PEDOT–C-MoS2 composite with DNA further confirms the presence of DNA; the sizes of the individual particles increased to approximately 222 nm. The EDS measurement also reports their presence in the hybrid nanostructure (Fig. 2F). | ||
| Fig. 2 SEM images of (A) α-Fe2O3 nanorods, (B) C-MoS2 nanosheets, (C) PEDOT nanospheres, (D) α-Fe2O3–PEDOT–C-MoS2 composite, (E) α-Fe2O3–PEDOT–C-MoS2–DNA hybrid nanostructure, (F) EDS spectrum of hybrid nanostructure. | ||
3.3 Electrochemical characterization
 | ||
| Fig. 3 (A) CV and (B) EIS profiles: (a) bare, (b) C-MoS2, (c) α-Fe2O3, (d) PEDOT, (e) α-Fe2O3–PEDOT–C-MoS2, (f) α-Fe2O3–PEDOT–C-MoS2–DNA modified electrodes at 50 mV s−1 in 0.1 M KCl and 1 mM [Fe(CN)6]3−/4−. | ||
The impedance behavior of the modified electrode recorded in the frequency region of 100 kHz to 1 Hz at an applied DC potential of 230 mV and an amplitude of ±5 mV is shown in Fig. 3B. The values of the charge transfer resistances (RCT) were determined from the Randles equivalent circuit. The RCT values for the α-Fe2O3–PEDOT–C-MoS2 composite, α-Fe2O3 bare GC, C-MoS2, PEDOT and α-Fe2O3–PEDOT–C-MoS2–DNA hybrid modified electrodes have been estimated as 117, 193, 229, 5341, and 8986 Ω, respectively. The obtained impedance plots were in good agreement with the CV measurements (Fig. 3A).
 | ||
| Fig. 4 CV: (A) (a) bare, (b) C-MoS2, (c) α-Fe2O3, (d) PEDOT, (e) α-Fe2O3–PEDOT–C-MoS2 composite, (f) α-Fe2O3–PEDOT–C-MoS2–DNA hybrid in 500 μM RF at 50 mV s−1, (B) pH versus current. | ||
Since electrons/protons participate in the redox chemistry of biomolecules, the redox peak current and potential are pH dependent. As shown in Fig. 4B, the α-Fe2O3–PEDOT–C-MoS2–DNA hybrid nanostructure system generated the maximal catalytic current at pH 7 and gradually decreased its catalytic activity when the pH was increased to neutral and basic values.
In comparison, a linear relationship of Ipc with scan rate1/2 was observed (Fig. 5A–C), suggesting that RF was electrocatalytically oxidized at α-Fe2O3–PEDOT–C-MoS2–DNA/GCE, α-Fe2O3–PEDOT–C-MoS2/GCE and α-Fe2O3–PEDOT–C-MoS2–DNA/SPE, respectively. This report confirms that all these electrode processes are diffusion-controlled.
 | ||
| Fig. 5 (A) CVs obtained for RF 100 μM: (A) α-Fe2O3–PEDOT–C-MoS2/GCE, (B) α-Fe2O3–PEDOT–C-MoS2–DNA/GCE, (C) α-Fe2O3–PEDOT–C-MoS2–DNA/SPE modified electrode recorded in PBS (pH 7.0) at different scan rates of 10 to 100 mV s−1. | ||
Fig. S2† shows the linear sweep voltammogram (LSV) obtained for RF sensing with different concentration ranges at α-Fe2O3–PEDOT–C-MoS2/GCE and α-Fe2O3–PEDOT–C-MoS2–DNA/GCE. The oxidation current of RF increased with a slight potential shift with increasing scan rate upon each increment of analyte. The oxidation currents had linear relationships with the concentration of RF, with correlation coefficients of 0.9849 and 9890, respectively (inset of Fig. S2†).
3.4 Square wave voltammetry on RF determination
On α-Fe2O3–PEDOT–C-MoS2/GCE, the SWV (Square Wave Voltammetry) oxidation peak potential of RF was at −0.486 V, and the oxidation peak currents of the analyte increased linearly from 2.890 × 10−4 A to 4.476 × 10−4 A with increasing concentration. Linear responses for RF determination were observed in the concentration range of 800 nM to 700 μM with a detection limit of 79 nM (S/N = signal/noise = 3σ/b, σ – standard deviation and b slope of the linear fit, Fig. 6). The greater gap of the interlayer distance in C-MoS2 compared with bulk MoS2 can provide desirable channels for electron transfer with reduced diffusion barriers.49 Meanwhile, on α-Fe2O3–PEDOT–C-MoS2–DNA/GCE, the peak RF potential was at −0.458 V and the oxidation peak currents were from 2.841 × 10−6 A to 4.482 × 10−5 A with increased concentration. The linear responses for this modified electrode were in the range of 300 nM to 1 mM, with a detection limit of 5 nM (Fig. 7). Two points have been noted from the above discussion: (i) the DNA modified electrode reduces the oxidation potential of RF by 30 mV, clearly showing electrocatalytic behavior. (ii) However, the oxidation peak current values decreased in the DNA modified electrode by 2 orders on the lower concentration side and one order in the higher concentration side in comparison with the modified electrode without DNA. Interestingly, the interaction between ferrites and phosphate present in DNA in the composite provides excellent electrocatalytic activity towards the oxidation of RF.50 On α-Fe2O3–PEDOT–C-MoS2–DNA/SPE, the SWV oxidation peak potential of RF is at −0.588 V and linear responses for the determination of RF were observed in the concentration range of 100 nM to 1 mM, with a detection limit of 12 nM (Fig. 8). The oxidation peak current values were from 2.267 × 10−6 A to 6.201 × 10−5 A. It is clear from the above 3 SWV results that (i) the DNA modified electrode detects a wider range than the other electrodes; (ii) even though the electroactive surface area (8.15 ×10−6 cm−2) of the α-Fe2O3–PEDOT–C-MoS2/GCE is higher, the sensitivity is not satisfactory, whereas the α-Fe2O3–PEDOT–C-MoS2–DNA/GCE (2.11 × 10−5 cm−2) and α-Fe2O3–PEDOT–C-MoS2–DNA/SPE (5.27 × 10−5 cm−2) possess comparatively less area, but exhibit more sensitivity and wide detection ranges. It is believed that the DNA in the hybrid nanostructure oxidizes the RF by a lower potential of 30 mV, which is responsible for the enhanced detection range. The corresponding detection limits and response ranges of different modified electrodes are listed (Table S4†) for comparison. Therefore, this hybrid sample, as an advanced electrode material, may have promising applications in electrochemical detection. The following statements suggest reasons for the reduced conductivity of the Fe2O3–PEDOT–C-MoS2–DNA hybrid material: | ||
| Fig. 6 SWV profiles: (A) 800 nM to 700 μM RF. (B) Plot of the oxidation peak current against the concentration of RF at α-Fe2O3–PEDOT–C-MoS2 composite/GCE in 0.1 M PBS (pH 7.0). | ||
 | ||
| Fig. 7 SWV profiles: (A) 300 nM to 1 mM RF. (B) Plot of the oxidation peak current against the concentration of RF at α-Fe2O3–PEDOT–C-MoS2–DNA hybrid nanostructure/GCE in 0.1 M PBS (pH 7.0). | ||
 | ||
| Fig. 8 SWV profiles: (A) 100 nM to 1 mM RF. (B) Plot of the oxidation peak current against the concentration of RF at α-Fe2O3–PEDOT–C-MoS2–DNA hybrid nanostructure/SPE in 0.1 M PBS (pH 7.0). | ||
(i) In DNA, the nucleobases are buried between the densely negatively charged phosphate backbones, and the DNA stayed away from the basal plane of C-MoS2, resulting in weak interaction.51
(ii) The affinity of MoS2 toward ds-DNA is lower than toward ss-DNA.52
(iii) Strong interactions between DNA and RF could be responsible for possible damage or shielding of the oxidizable groups of guanine and adenine bases present in DNA.53
However, a strong interaction/fast formation of PEDOT and DNA in the hybrid due to electrostatic attraction between the phosphate groups of DNA and PEDOT may result in the enhanced electrocatalytic activity.54 Moreover, it is believed that in the PEDOT–DNA system, the increased conductivity is due to the modulation ability of the DNA conformation in the redox process, which also supports the enhanced catalytic behavior. Hence, the design of this scaffold with DNA could be used for the fabrication of a nanostructured catalyst for biosensing applications.
3.5 Reproducibility and stability of sensors
The reproducibility of the α-Fe2O3–PEDOT–C-MoS2–DNA/GCE was investigated by the SWV responses of RF. The relative standard deviation (RSD) responses for six independent electrodes were 5.4%. The fabricated sensors with α-Fe2O3–PEDOT–C-MoS2/GCE, α-Fe2O3–PEDOT–C-MoS2–DNA/GCE and α-Fe2O3–PEDOT–C-MoS2–DNA/SPE were subjected to 100 continuous cycles in the presence of 1 mM RF (Fig. S3†). The long-term stability of the electrode stored at 4 °C was measured after one month of storage in dry conditions. The electrode retained 88% of its initial response. These results demonstrate that the modified electrodes possessed satisfactory reproducibility and tremendous stability for analytical applications.3.6 Practical applications
The anti-interference ability of the α-Fe2O3–PEDOT–C-MoS2–DNA/GCE was examined by the addition of several types of ions and other small molecules to PBS solution containing 100 μM RF (Fig. S4 and Table S1†). No interference was observed in the presence of 10 fold excesses of ascorbic acid, KCl, FeCl3, folic acid, MgCl3, uric acid and tyrosine. It is clear that the proposed sensor exhibited good selectivity for the determination of RF. The utilization of the α-Fe2O3–PEDOT–C-MoS2–DNA/GCE sensor in real samples was also studied by the standard addition method in human urine, milk powder and pharmaceutical drug samples. All the details of the real sample analysis results are summarized in Tables S2 and S3.†The urine sample was diluted 10 times with 0.1 M PBS solution (pH 7.0) without any other treatment before measurement. While adding the diluted sample, a sharp peak appeared for RF at 0.42 V for 10 μM concentration. For different concentrations (10 to 80 μM) of urine sample, the corresponding RF peaks were recorded and are shown in Fig. S5(A).† The same experiment was performed with milk powder, and a peak appeared at 0.48 V for 100 μM. The responses for different concentrations (100 μM to 1 mM) are shown in Fig. S5(B).† Finally, the RF oxidation of the pharmaceutical drug was analyzed at 0.41 V for 100 μM. The square wave studies for different concentrations of the drug (10 μM to 100 μM) are depicted in Fig. S5(C).†
4. Conclusions
A simple chemical route has been reported for the synthesis of α-Fe2O3–PEDOT–C-MoS2–DNA hybrid nanostructures. The fabricated α-Fe2O3–PEDOT–C-MoS2–DNA modified electrodes, which integrate the unique behavior of the components (α-Fe2O3 has enzymatic activity, C-MoS2 mimics the activity of graphene, and DNA has catalytic activity), provided superior currents for the analyte RF compared to those of α-Fe2O3–PEDOT–C-MoS2/GCE. It is demonstrated that the DNA modified composite exhibits a significantly low detection limit and high electrocatalytic activity. Additionally, based on its electrochemical performance, the DNA modified hybrid structure may be a platform with high potential in biomedical and non-corrosive device fabrication.Acknowledgements
The author J. W. gratefully acknowledges the University Grant Commission (MRP-MAJOR-ELEC-2013-37628) for financial assistance.References
- G. Gastaldi, G. Ferrari, A. Verri, D. Casirola, M. N. Orsenigo and U. J. Laforenza, Nutrition, 2000, 130, 2556–2561 Search PubMed.
- D. W. Bat and C. D. Anal Eckhert, Biochemistry, 1990, 188, 164–167 Search PubMed.
- S. A. Berhe, J. Y. Zhou, K. M. Haynes, M. T. Rodriguez and W. J. Youngblood, ACS Appl. Mater. Interfaces, 2012, 4, 2955–2963 Search PubMed.
- S. A. Carter, Appl. Phys. Lett., 1997, 71, 1145–1147 Search PubMed.
- R. Brown, C. P. Jarrett, D. M. Leeuw and M. Matters, Synth. Met., 1997, 88, 37–55 CrossRef.
- A. Pron and P. Rannou, Prog. Polym. Sci., 2002, 27, 135–190 CrossRef CAS.
- G. U. O. Xian-zhi, K. Yan-fei, Y. Tai-li and W. Shu-rong, Trans. Nonferrous Met. Soc. China, 2012, 22, 380–385 CrossRef.
- Y. Wang, W. Jia, T. Strout, A. Schempf, H. Zhang, B. Li and J. Cui, Electroanalysis, 2009, 21, 1432–1438 CrossRef CAS.
- M. D. McGehee and J. Heeger, Adv. Mater., 2000, 12, 1655–1668 CrossRef CAS.
- H. Kim, N. Schulte, G. Zhou, K. Müllen and F. Laquai, Adv. Mater., 2011, 23, 894–897 CrossRef CAS PubMed.
- J. J. Davis, M. L. H. Green, H. A. O. Hill, Y. C. Leung, P. J. Sadler, J. Sloan, A. V. Xavier and S. C. Tsang, Inorg. Chim. Acta, 1998, 272, 261 CrossRef CAS.
- M. Wang, F. Zhao, Y. Liu and S. Dong, Biosens. Bioelectron., 2005, 21, 159 CrossRef CAS PubMed.
- G. Heywang and F. Jonas, Adv. Mater., 1992, 4, 116 CrossRef CAS.
- H. Yamato, M. Ohwa and W. Wernet, J. Electroanal. Chem., 1995, 397, 163 CrossRef.
- S. Radhakrishnan, C. Sumathi, V. Dharuman and J. Wilson, Anal. Methods, 2013, 5, 684–689 RSC.
- A. A. Ansari, P. R. Solanki and B. D. Malhotra, Appl. Phys. Lett., 2008, 92, 263901 CrossRef.
- J. Liu, Y. Li, X. Huang and Z. Zhu, Nanoscale Res. Lett., 2010, 5, 1177 CrossRef CAS PubMed.
- H. J. Kim, S. H. Yoon, H. N. Choi, Y. K. Lyu and W. Y. Lee, Bull. Korean Chem. Soc., 2006, 27, 65 CrossRef CAS.
- R. Doong and H. Shih, Biosens. Bioelectron., 2010, 25, 1439 CrossRef CAS PubMed.
- M. Thandavarayan, J. Sundaramurthy, P. Suresh Kumar, P. Kannan, O. Marcin and S. Ramakrishna, Analyst, 2013, 138, 1779 RSC.
- C. Sumathi, C. Venkateswara Raju, P. Muthukumaran, J. Wilson and G. Ravi, J. Mater. Chem. B, 2016, 4, 2561–2569 RSC.
- H. S. S. R. Matte, A. Gomathi, A. K. Manna, D. J. Late, R. Datta, S. K. Pati and C. N. R. Rao, Angew. Chem., Int. Ed., 2010, 49, 4059–4062 CrossRef CAS PubMed.
- H. Hwang, H. Kim and J. Cho, Nano Lett., 2011, 11, 4826–4830 CrossRef CAS PubMed.
- J. M. Soon and K. P. Loh, Electrochem. Solid-State Lett., 2007, 10, A250–A254 CrossRef CAS.
- J. Chen, N. Kuriyama, H. T. Yuan, H. T. Takeshita and T. Sakai, J. Am. Chem. Soc., 2001, 123, 11813–11814 CrossRef CAS PubMed.
- W. H. Hu, X. Shang, G. Q. Han, B. Dong, Y. R. Liu, X. Li, Y. M. Chai, Y. Q. Liu and C. G. Liu, Carbon, 2016, 100, 236–242 CrossRef CAS.
- W. H. Hu, G. Q. Han, Y. R. Liu, B. Dong, Y. M. Chai, Y. Q. Liu and C. G. Liu, Int. J. Hydrogen Energy, 2015, 40, 6552–6558 CrossRef CAS.
- T. Scheibel, R. Parthasarathy, G. Sawicki, X. M. Lin, H. Jaegerand and S. L. Lindquist, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4527 CrossRef CAS PubMed.
- S. W. Lee, C. B. Mao, C. E. Flynn and A. M. Belcher, Science, 2002, 296, 892 CrossRef CAS PubMed.
- R. A. Mcmillan, C. D. Paavola, J. Howard, N. J. Zaluzec and J. D. Trent, Nat. Mater., 2002, 1, 24 Search PubMed.
- R. Bhandari, D. B. Pacardo, N. M. Bedford, R. R. Naik and M. R. Knecht, J. Phys. Chem. C, 2013, 117, 18053 CAS.
- C. A. Mirkin, Inorg. Chem., 2000, 39, 2258 CrossRef CAS PubMed.
- L. Berti, A. Alessandrini and P. Facci, J. Am. Chem. Soc., 2005, 127, 11216 CrossRef CAS PubMed.
- K. Keren, M. Krueger, R. Gilad, G. Ben-Yoseph, U. Sivan and E. Braun, Science, 2002, 297, 72 CrossRef CAS PubMed.
- C. F. Monson and A. T. Woolley, Nano Lett., 2003, 3, 359 CrossRef CAS.
- R. Seidel, L. C. Ciacchi, M. Weigel, W. Pompe and M. Mertig, J. Phys. Chem. B, 2004, 108, 10801 CrossRef CAS.
- H. Wang, P. Chen, F. Wen, Y. Zhu and Y. Zhang, Sens. Actuators, B, 2015, 220, 749–754 CrossRef CAS.
- F. Terzi, L. Pasquali, M. Montecchi, S. Annarone, A. Viinikanoja, T. Aritalo, M. Salom aki and J. Lukkari, J. Phys. Chem. C, 2011, 115, 17836–17844 CAS.
- M. Sevilla and A. B. Fuertes, Carbon, 2009, 47, 2281 CrossRef CAS.
- D. B. Nimbalkar, H. H. Lo, P. V. R. K. Ramacharyulu and S. C. Ke, RSC Adv., 2016, 6, 31661–31667 RSC.
- Y. P. He, Y. M. Miao, C. R. Li, S. Q. Wang, L. Cao and S. S. Xie, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 125411 CrossRef.
- C. J. Sartoretti, B. D. Alexander, R. Solarska, I. A. Rutkowska, J. Augustynski and R. Cerny, J. Phys. Chem. B, 2005, 109, 13685 CrossRef CAS PubMed.
- L. Zhang, R. Jamal, Q. Zhao, M. Wang and T. Abdiryim, Nanoscale Res. Lett., 2015, 10, 148 CrossRef PubMed.
- S. H. Jo, Y. K. Lee, J. W. Yang, W. G. Jung and J. Y. Kim, Synth. Met., 2012, 162, 1279–1284 CrossRef CAS.
- C. Sumathi, P. Muthukumaran, S. Radhakrishnan, G. Ravi and J. Wilson, RSC Adv., 2015, 5, 17888–17896 RSC.
- K. Chang, W.-X. Chen, L. Ma, H. Li, H. Li, F. Huang, Z. Xu, Q. Zhang and J.-Y. Lee, J. Mater. Chem., 2011, 21, 6251 RSC.
- D. Kong, H. Wang, J. J. Cha, M. Pasta and K. J. Ko ski, Nano Lett., 2013, 13, 1341 CrossRef CAS PubMed.
- J. Wang, Z. Wua, H. Yinb, W. Li and Y. Jiang, RSC Adv., 2014, 4, 56926–56932 RSC.
- J. Xiao, D. Choi, L. Cosimbescu, P. Koech, J. Liu and J. P. Lemmon, Chem. Mater., 2010, 22, 4522–4524 CrossRef CAS.
- P. Dai and Z. Yang, Microchim. Acta, 2012, 176, 109–115 CrossRef CAS.
- B. L. Li, H. L. Zou, L. Lu, Y. Yang, J. L. Lei, H. Q. Luo and N. Bing, Adv. Funct. Mater., 2015, 25, 3541–3550 CrossRef CAS.
- K. K. Zadeh and J. Z. Ou, ACS Sens., 2016, 1, 5–16 CrossRef.
- A. A. Ensafi, E. Heydari-Bafrooei and M. Amini, Biosens. Bioelectron., 2012, 31, 376–381 CrossRef CAS PubMed.
- Y. Ner, M. A. Invernale, J. G. Grote, J. A. Stuart and G. A. Sotzing, Synth. Met., 2010, 160, 351–353 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra16279g |
| This journal is © The Royal Society of Chemistry 2016 |