
Organoaluminum(III) complexes of the bis-N,N′-(2,6-diisopropylphenyl)imidazolin-2-imine ligand†
Angela D. K. Todd,
William L. McClennan and
Jason D. Masuda*
The Atlantic Centre for Green Chemistry, Department of Chemistry, Saint Mary's University, Halifax, Nova Scotia, Canada B3H 3C3. E-mail: jason.masuda@smu.ca; Tel: +1-902-420-5077
First published on 18th July 2016
Abstract
The reaction of trimethylaluminum with the bis-N,N′-(2,6-diisopropylphenyl)imidazolin-2-imine (L–H) ligand 1 afforded several new organometallic aluminium complexes. Reaction in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry at room temperature afforded a Lewis acid–base adduct, whereas thermolysis resulted in the loss of methane and the formation of a dimer complex, (L-AlMe2)2, 3. When reacted in a 1:2 ratio at 110 °C, the loss of two equivalents of methane yielded L2AlMe, 5, whereas when the reaction was performed at 60 °C, (L–H)AlMe2(L), 4, was found as an intermediate in the reaction. Compound 2 is, to the best of our knowledge, the first example of a structurally characterized primary imine coordinated to a triorganoaluminum(III) center. Attempts to form a two coordinate aluminum cation by CH3− abstraction are documented. All products were characterized via normal spectroscopic techniques and single crystal X-ray diffraction.
:1 stoichiometry at room temperature afforded a Lewis acid–base adduct, whereas thermolysis resulted in the loss of methane and the formation of a dimer complex, (L-AlMe2)2, 3. When reacted in a 1:2 ratio at 110 °C, the loss of two equivalents of methane yielded L2AlMe, 5, whereas when the reaction was performed at 60 °C, (L–H)AlMe2(L), 4, was found as an intermediate in the reaction. Compound 2 is, to the best of our knowledge, the first example of a structurally characterized primary imine coordinated to a triorganoaluminum(III) center. Attempts to form a two coordinate aluminum cation by CH3− abstraction are documented. All products were characterized via normal spectroscopic techniques and single crystal X-ray diffraction.
Introduction
Within the last several decades, the amido-based family of ligands including guanidinate [R2NC(NR′)2]− systems have been extensively studied, primarily as ancillary ligands in the area of α-olefin polymerization in regards to catalytically active group 4 metal complexes.1–3 Historically these molecules have functioned as building blocks in natural products4–6 and have been utilized in the formation of pharmaceutical agents but they have garnered increasing attention in the field of small molecule activation.7–9In addition to the synthesis of several group 4 catalysts, many series of lanthanide and actinide complexes have been reported for the use in effective homogeneous catalysts7,10,11 and precursors for rare-earth oxide thin films.7,12 The success of these ligands results mainly from their steric tunability and efficient π-donation that often exceeds conventional cyclopentadienyl ligands.13
Of particular interest is the closely related imidazolin-2-iminato family of ligands first developed by Kuhn et al.,14 of which even, stronger electron donating capacity has been observed.15,16 They have shown particular success as ancillary ligands in titanium-based olefin polymerization catalysis.13,17–25 Due to the nature of these particular ligands, as anions, they can function as 2σ,2π-electron donors or as 2σ,4π-electron donors in a zwitterion form; these are effective at stabilizing early transition metal complexes or those of high oxidation states due to an increased contribution of the more polarized zwitterion shown in Scheme 1.26,27 This feature has made this ligand a versatile replacement as it interacts in as similar fashion to phosphinimides, R3PN−,28–30 and therefore can be utilized as monodentate alternatives to Cp-based ligands and has shown additional developments in the applications of catalytic dimerization of aldehydes,31 lanthanide-based hydroamination catalysts,15,26,32 ruthenium-based transfer hydrogenation33–35 as well as copper36 and palladium chemistry.37,38
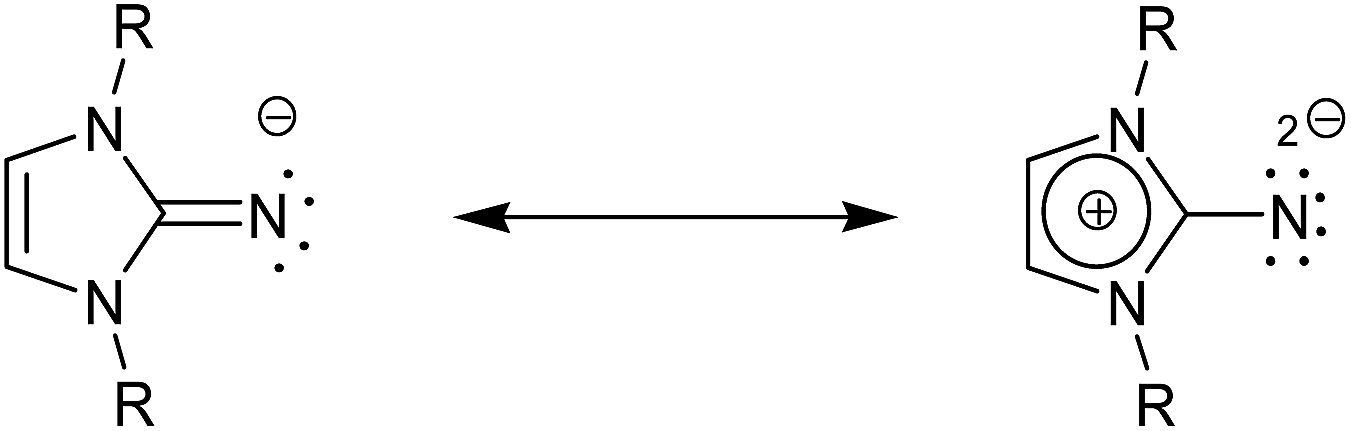 | ||
| Scheme 1 Mesomeric forms of imidazolin-2-iminato ligands. | ||
Recently main group chemists have utilized the exceptional donating and steric properties of the title ligand to stabilize unique molecular frameworks. This is highlighted by the group 15 chemistry of Bertrand with the isolation of a carbene stabilized phosphorus mononitride,39 a monomeric phosphinyl radical,40 a singlet phosphinonitrene,41 its protonated derivative42 and metal complexes.43 The imidazolin-2-iminato ligand has also been used by Inoue44–48 and Rivard49,50 to stabilize a number of group 13- and group 16-based molecules, in particular the remarkable isolation of the first molecule containing an Al![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Te double bond.46 The organo-aluminum chemistry of the title imidiazoline-2-iminato ligand is relatively understudied and has focussed on the aluminum hydrides.47,48
Te double bond.46 The organo-aluminum chemistry of the title imidiazoline-2-iminato ligand is relatively understudied and has focussed on the aluminum hydrides.47,48
Our research group has had a long standing interest in the stabilization of unusual main group compounds.51,52 These include the isolation and reactivity studies of a phosphinyl radical,53,54 as well as the isolation of the first oxophosphonium cation.55 Similarly, we have been interested in the isolation of low-valent and low-oxidation state aluminium complexes. Herein, we explore the reactivity of the bis-N,N′-(2,6-diisopropylphenyl)imidazolin-2-imine ligand (L–H, 1) with trimethylaluminum in our quest of isolating new aluminium cations and aluminium(I) species. We report the isolation of a number of compounds that have been characterized by various methods including NMR spectroscopy and single crystal X-ray crystallography.
Results and discussion
Ligand 1 was reacted with trimethylaluminum in pentane at room temperature to give compound 2 as a beige colored powder in high yield (Scheme 2). The 1H NMR spectrum revealed a singlet at δ −0.81 ppm that integrated to 9 hydrogen atoms and was assigned to the three methyl groups on aluminum. A broad singlet at δ 4.07 ppm was assigned to the imine proton. Combined, these indicated that a simple Lewis acid–base adduct was formed with the between aluminum and the imine nitrogen with no deprotonation of the imine. Although the molecule would be expected to have low symmetry, the presence of two doublets and a single septet related to the iPr groups indicate a dynamic process occurring at room temperature averaging out these signals. Presumably this is rotation about the imine N–C bond, implying that there is a reduced bond order between these atoms.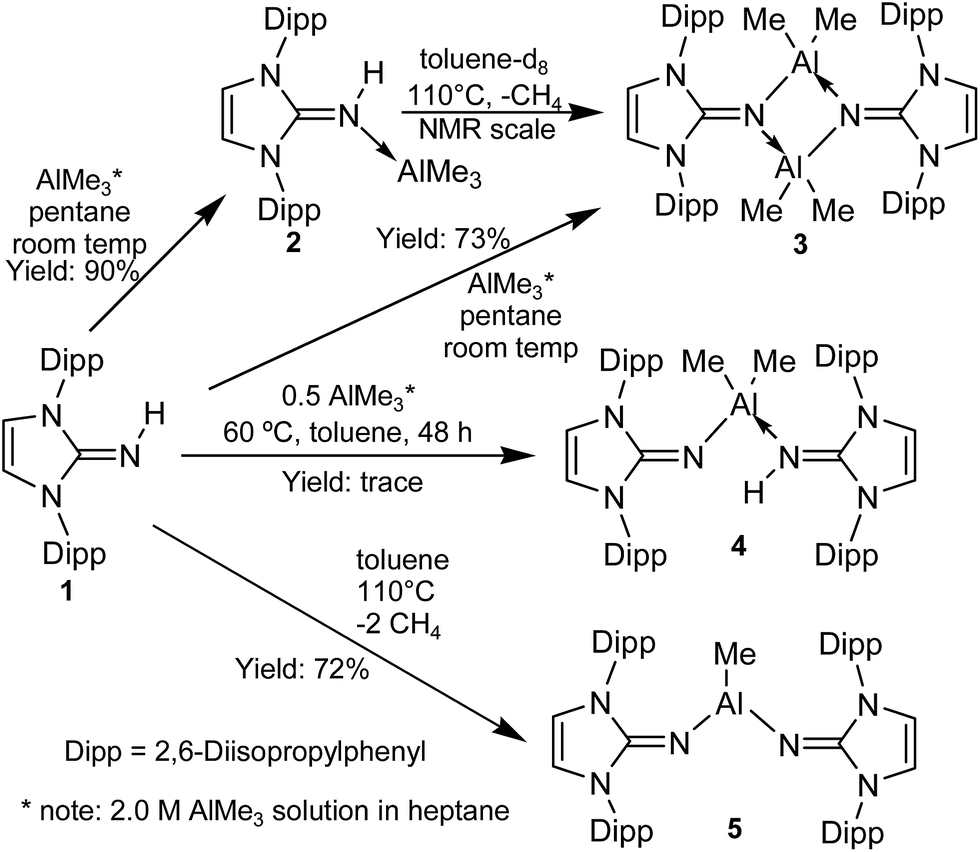 | ||
| Scheme 2 Reactions of ligand 1 with trimethylaluminum. | ||
A Cambridge Structural Database search (CSD version 5.37 (April 2015)) reveals that this is the first structurally characterized primary imine coordinated to a triorganoaluminum(III) center (Fig. 1). The Al–N distance (1.9648(19) Å) is longer than nitrogen–AlMe3 adducts such as Ph3PN(H)AlMe3 (1.672(4) Å)56 and slightly shorter than the N–Al bond in the ammonia–AlMe3 adduct (2.004(5) Å).57 The imine NC distance is 1.313(3) Å, slightly elongated compared to the parent imine with an NC distance of 1.2888(17) Å.58 The C1–N2 and C1–N3 distances (1.366(2) Å and 1.366(3) Å, respectively) are shorter than in the parent ligand (1.3815(15) Å, 1.4004(16) Å). This can be attributed to a significant contribution from the zwitterionic structure as shown in Scheme 1. The 1:1 reaction between ligand 1 and trimethylaluminum at elevated temperature (110 °C) in toluene gave compound 3 at moderate yields. Alternatively, the NMR-scale reaction of heating compound 2 to 50 °C in toluene-d8 resulted in the slow formation of compound 3 and methane. The 1H NMR spectrum revealed a peak at δ −1.50 ppm that integrated to six hydrogen atoms, confirming the loss of a methyl group. This along with the loss of the imine N–H signal supported the formation of a covalent bond between the nitrogen and aluminum.
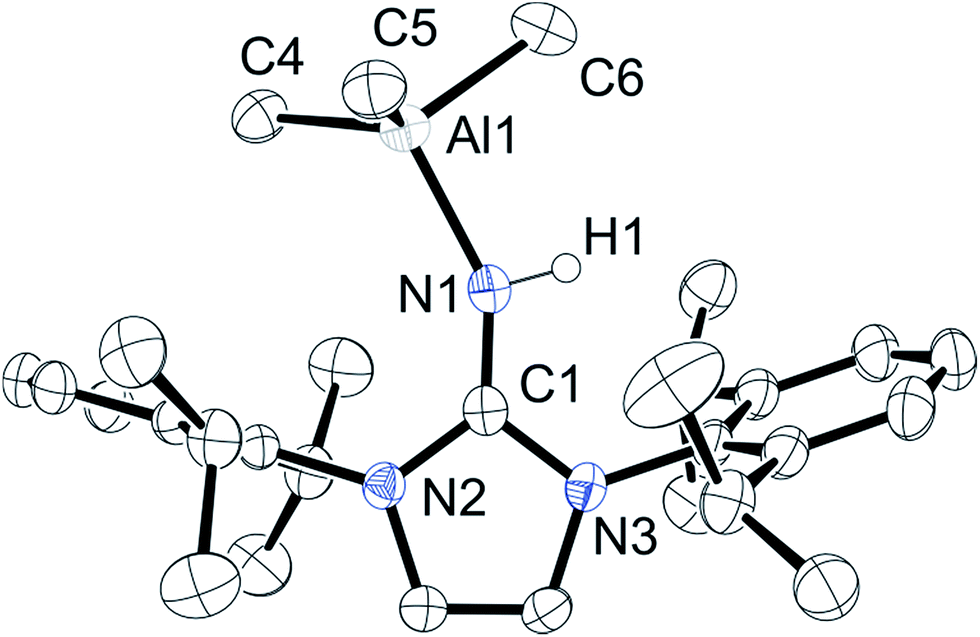 | ||
| Fig. 1 Molecular structure of compound 2, with thermal ellipsoids projected at the 50% probability level. Co-crystallized toluene and hydrogen atoms (except H1) have been omitted for clarity. Selected bond lengths (Å) and angles (°): Al1–N1 1.9648(19), N1–C1 1.313(3), N1–H1 0.86(2), N2–C1 1.366(2), N2–C2 1.400(3), N3–C1 1.366(3), N3–C3 1.394(2), C2–C3 1.332(3). | ||
Recrystallization of the product from hot heptane cooled to ambient temperature afforded crystals suitable for single crystal X-ray diffraction. This compound crystallized in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group and revealed a dimerized product (Fig. 2). The four-membered Al–N–Al–N is planar; the N3–Al1–N6 and N3–Al2–N6 angles are 86.6(1)° and the Al1–N3–Al2 and Al1–N6–Al2 angles are 93.3(1)°. The CN imine distances for C1–N3 (1.292(4) Å) and C28–N6 (1.302(4) Å) distances are statistically indistinguishable from the imine distance in 2, indicating the important contribution of the zwitterionic form of the ligand (Scheme 1).
space group and revealed a dimerized product (Fig. 2). The four-membered Al–N–Al–N is planar; the N3–Al1–N6 and N3–Al2–N6 angles are 86.6(1)° and the Al1–N3–Al2 and Al1–N6–Al2 angles are 93.3(1)°. The CN imine distances for C1–N3 (1.292(4) Å) and C28–N6 (1.302(4) Å) distances are statistically indistinguishable from the imine distance in 2, indicating the important contribution of the zwitterionic form of the ligand (Scheme 1).
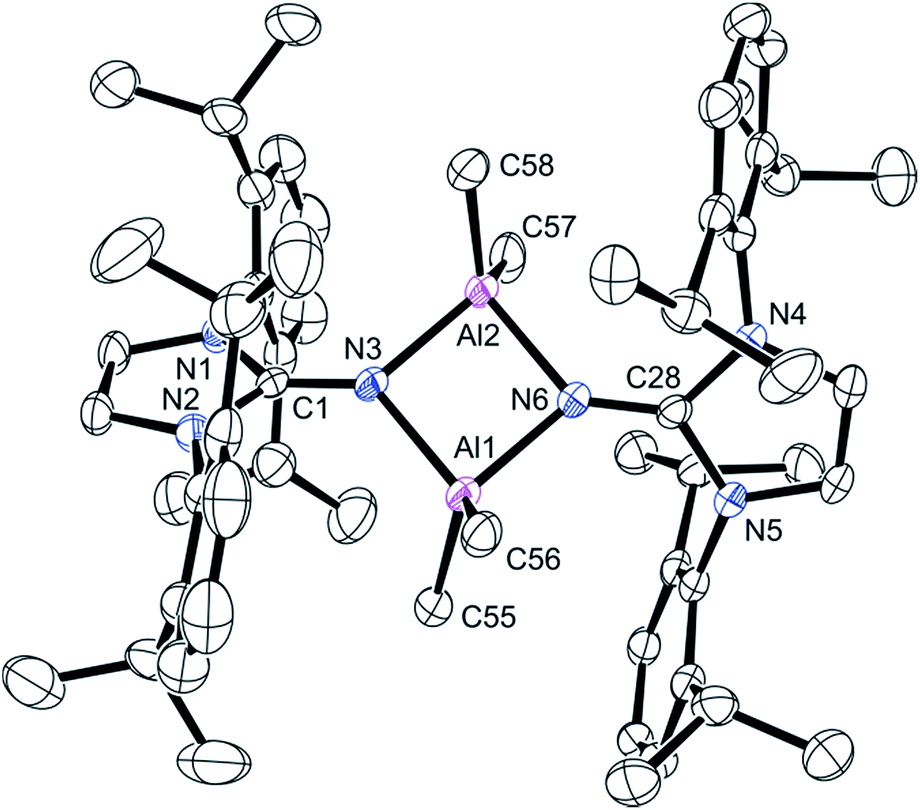 | ||
| Fig. 2 Molecular structure of compound 3, with thermal ellipsoids projected at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): Al1–N3 1.919(3), Al1–N6 1.923(2), Al2–N3 1.924(2), Al2–N6 1.919(3), N3–C1 1.292(4), N6–C28 1.302(4), N2–C1 1.403(3), C2–C3 1.329(5), C29–C30 1.322(5), N3–Al1–N6 86.6(1), N3–Al2–N6 86.6(1), Al1–N3–Al2 93.3(1), Al1–N6–Al2 93.3(1). | ||
The reaction of ligand 1 in a 2:1 ratio with trimethylaluminum at 60 °C in toluene for two days gave a mixture of products as determined by 1H NMR spectroscopy. A small amount of crystalline material was isolated from a hot heptane solution cooled to room temperature. Analysis by single crystal X-ray diffraction revealed compound 4 crystallized as neutral and anionic ligand 1 coordinated to an AlMe2 fragment (Fig. 3). The asymmetric nature of this compound is apparent in the aluminium–nitrogen distances; the adduct side had an Al1–N4 distance of 1.965(2) Å whereas the covalently bound ligand has a much shorter Al1–N1 distance of 1.822(2) Å. There is also a marked difference in the NC imine bonds; the covalently bound ligand has an N4–C6 imine bond length of 1.313(3) Å, whereas the covalently bound ligand has a much shorter N1–C3 imine bond length of 1.257(3) Å. An attempt to prepare 4 by adding one equivalent of ligand 1 to dimer 3, was unsuccessful. 1H NMR spectroscopy showed no reaction at room temperature. Heating this mixture to 110 °C resulted in the loss of signals of the starting materials and gave a new Al–Me signal in the 1H NMR spectrum at δ −1.49 ppm, integrating to three hydrogen atoms. This signal was assigned to compound 5.
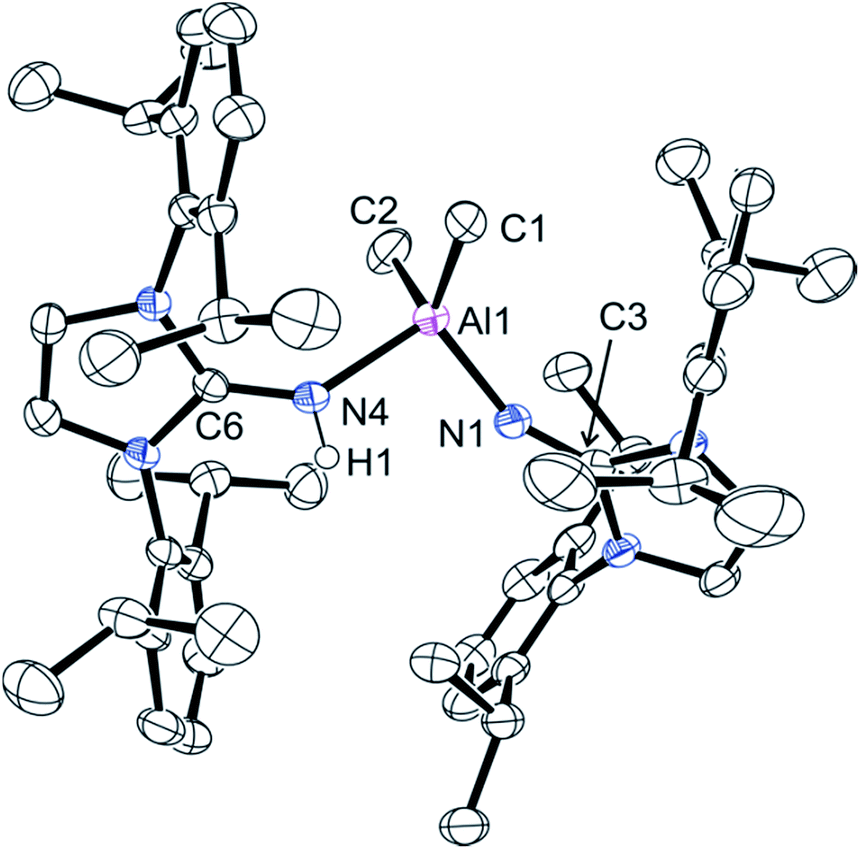 | ||
| Fig. 3 Molecular structure of compound 4, with thermal ellipsoids projected at the 50% probability level. Hydrogen atoms aside from H1 have been omitted for clarity. Selected bond lengths (Å) and angles (°): Al1–C1 1.967(2), Al1–C2 1.985(3), Al1–N1 1.822(2), Al1–N4 1.965(2), N1–C3 1.257(3), N4–C6 1.313(3), N4–H1 0.86(2), C1–Al1–C2 111.8(1), N1–Al1–N4 96.95(8), Al1–N1–C3 152.2(2), Al1–N4–C6 137.6(2). | ||
In a more direct approach, heating the 2:1 mixture of 2 and trimethylaluminum at 110 °C in toluene for two days gave a single product by 1H NMR spectroscopy. The loss of two methyl groups from the trimethylaluminum and additional loss of the imine protons and resulting highly symmetrical 1H NMR spectrum gave evidence towards the formation of 5. Recrystallization of the isolated product from hot heptane yielded beige crystals suitable for single crystal X-ray diffraction. The product was found packed into a P21/c space group and the formation of a complex between two coordinated ligands to trimethylaluminum was confirmed as shown in Fig. 4. The Al centre is trigonal planar with angles of N1–Al1–N4 119.73(7)°, N1–Al1–C1 117.77(8)° and N4–Al1–C1 122.45(8)°. Both imine carbon–nitrogen bonds (N1–C2 1.266(2) Å, N4–C29 1.265(2) Å) showed significant shortening compared to the neutral Lewis adducts. Compared to the synthesized adduct 2 (Al1–N1 1.965(2) Å) and dimer 3 (Al1–N3 1.919(3) Å and Al1–N6 1.923(2) Å), product 5 had significantly shorter Al–N distances of Al1–N1 1.759(2) Å and Al1–N4 1.762(1) Å, reinforcing the covalent nature of these molecules. The imine C–N bonds in 5 (N1–C2 1.266(2) Å and N4–C29 1.265(2) Å) were found to be shorter than that in the free ligand. This shortening of the CN imine bond indicates the stronger participation of the first mesomeric form shown as shown in Scheme 1.
 | ||
| Fig. 4 Molecular structure of compound 5, with thermal ellipsoids projected at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): Al1–N1 1.759(2), Al1–N4 1.762(1), Al1–C1 1.950(2), N1–C2 1.266(2), N4–C29 1.265(2), C2–N2 1.413(2), C2–N3 1.400(2), N1–Al1–N4 119.73(7), C1–Al1–N1 117.77(8), C1–Al1–N4 122.45(8), Al1–N1–C2 140.8(1), Al1–N4–C29 143.4(1). | ||
Recrystallization of the isolated product from hot heptane yielded beige crystals suitable for single crystal X-ray diffraction. The product was found packed into a P21/c space group and the formation of a complex between two coordinated ligands to trimethylaluminum was confirmed as shown in Fig. 4. The Al centre is trigonal planar with angles of N1–Al1–N4 119.73(7)°, N1–Al1–C1 117.77(8)° and N4–Al1–C1 122.45(8)°. Both imine carbon–nitrogen bonds (N1–C2 1.266(2) Å, N4–C29 1.265(2) Å) showed significant shortening compared to the neutral Lewis adducts. Compared to the synthesized adduct 2 (Al1–N1 1.965(2) Å) and dimer 3 (Al1–N3 1.919(3) Å and Al1–N6 1.923(2) Å), product 5 had significantly shorter Al–N distances of Al1–N1 1.759(2) Å and Al1–N4 1.762(1) Å, reinforcing the covalent nature of these molecules. The imine C–N bonds in 5 (N1–C2 1.266(2) Å and N4–C29 1.265(2) Å) were found to be shorter than that in the free ligand. This shortening of the CN imine bond indicates the stronger participation of the first mesomeric form shown as shown in Scheme 1.
With significant quantities of compound 5 in hand, we tested it as a starting material with a two-coordinate aluminium cation as a target. Reaction of 5 with I2 in toluene to exchange a methyl group with iodide (and loss of MeI) resulted in an intractable mixture. This is in contrast with the efficient exchange as seen with NacnacAlMe2.59 Attempted abstraction of the methyl group with the strong Lewis acid B(C6F5)3 resulted in no reaction (or noticeable Al–Me–B interactions) by 1H NMR spectroscopy. Thermolysis of the mixture resulted in no change. Similarly, there was no reactivity noted of 5 with the trityl borate [Ph3C][B(C6H2(m-CF3)2)4].
Adding triflic acid to a stirring mixture of 5 in pentane resulted in the evolution of a gas (presumably methane) with formation of a colourless precipitate. 1H NMR spectroscopy of the mixture showed loss of the methyl group, but otherwise was not diagnostic. Although we could not ascertain the exact composition of the bulk material, we were successful at obtaining a few single crystals from THF solutions. Analysis by single crystal X-ray crystallography (compound 6, Fig. 5A) revealed a cation containing two equivalents of ligand 1 forming a dimer that is hydrogen bound to a central proton (no aluminum). More interesting is the anion; the central aluminum atom is in an octahedral configuration with two O-bound THF molecules in the axial positions and the four triflate ligands in the equatorial positions, each bound in a monodentate fashion. This is a rare example of aluminum containing four triflate anions acting as ligands; the only other example, [Al(triglyme)3][Al(OH2)2OTf4], was recently reported.60 From another reaction, another crystal was also analysed and was determined to be [1-H][OTf] (compound 7, Fig. 5B). In this compound, two of the oxygens of the triflate are hydrogen bound to the NH2+ fragment of the cation, forming with donor–acceptor bond distances and N–H⋯O angles of 2.890(3) Å and 125(3)°, and 2.868(3) Å and 144(3)° for each.
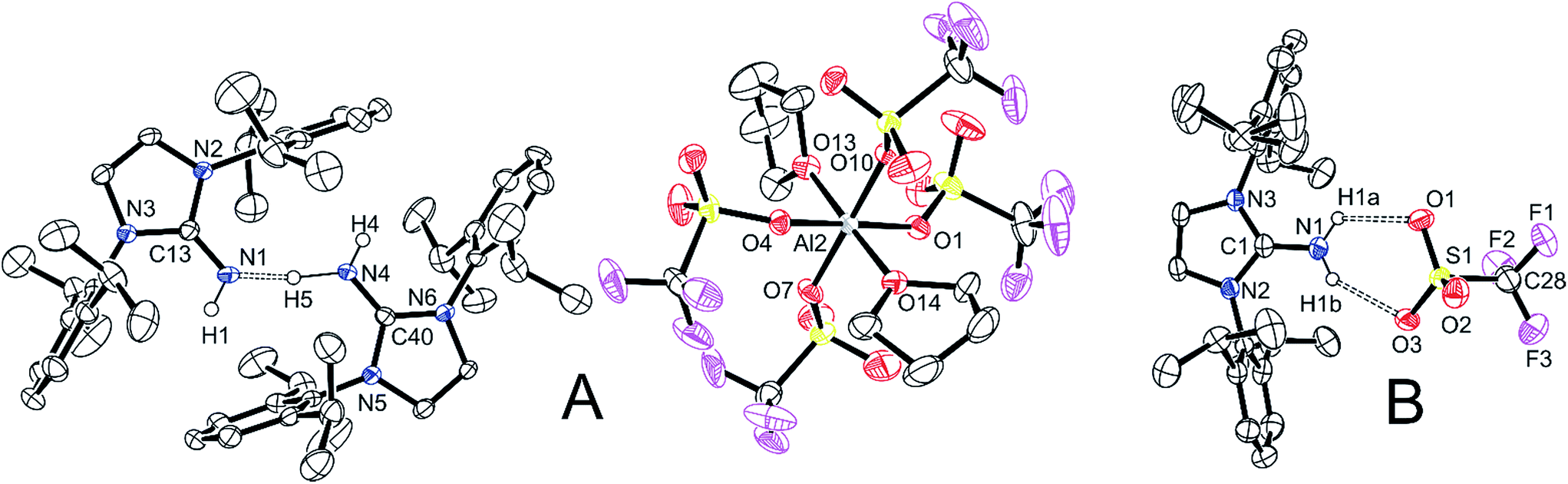 | ||
| Fig. 5 Molecular structure of compounds 6 (A) and 7 (B), with thermal ellipsoids projected at the 50% probability level. | ||
Conclusions
In summary, a novel class of aluminium complexes based from the imidazolin-2-iminato ligand have been successfully synthesised and completely characterized via a number of methods including NMR spectroscopy, EA and single crystal X-ray diffraction. To the best of our knowledge, compound 2 is the first example of structurally characterized primary imine coordinated to a triorganoaluminum centre. Compound 5 was resistant to methyl abstraction reactions using highly Lewis acidic B(C6F5)3 and trityl borates. Finally, addition of triflic acid to 5 resulted in the loss of Al–N bonds to form new products where ligand 1 acts as a Brønsted–Lowry base.Experimental
General synthetic procedures
All reactions were performed in dry, O2-free conditions under an atmosphere of N2 within an mBraun Labmaster SP inert atmosphere drybox or sealed reaction vessels using standard Schlenk techniques. Synthesis of ligand 1 was done via literature procedure published by Tamm et al.16 modified from the original synthesis by Kuhn et al.14 All reagents were purchased from Sigma-Aldrich and used as received, unless otherwise noted. Alumina and molecular sieves were pre-dried in a 150 °C oven before bring dried at 300 °C in vacuo. Solvents were purified using an Innovative Technology solvent purification system or purchased as ‘anhydrous’ from Sigma-Aldrich. Solvents were then dried using KH and subsequently filtered through dry alumina and stored over previously dried 3 Å molecular sieves. Glassware was dried at 150 °C overnight prior to experimentation. NMR spectra were recorded on a Bruker Avance 300 MHz spectrometer. Trace amounts of non-deuterated solvent were used as internal references for 1H NMR spectra and were referenced relative to tetramethylsilane. The deuterated solvent was used as an internal reference for 13C {1H} NMR spectra and referenced relative to tetramethylsilane. Coupling constants are reported as absolute values. Melting points were recorded on an Electrothermal MEL-Temp 3.0 using glass capillaries sealed under inert conditions. Elemental analysis was performed by the Centre for Environmental Analysis and Remediation (CEAR) facility at Saint Mary's University using a Perkin Elmer 2400 II series Elemental Analyser.Preparation of (L–H)-AlMe3 (2)
In a 100 mL round bottomed flask, ligand 1 (5.00 g, 12.4 mmol) was added to 50 mL pentane. 6.19 mL of a 2.0 M solution of trimethylaluminum in heptane (0.893 g, 12.4 mmol) was added to the resulting slurry. This mixture was then allowed to stir over 48 hours at ambient temperature. The solvent was subsequently removed in vacuo yielding the desired product as a beige solid pure by 1H NMR (yield: 5.33 g, 90%). Analytically pure crystals were recovered by a second recrystallization in ambient temperature toluene layered with pentane. Mp 178 °C (decomp.); anal. calc. for C30H46N3Al: C, 75.75; H, 9.75; N, 8.83%; found: C, 75.45; H, 9.68; N, 8.62%. 1H NMR (300 MHz, C6D6): δ −0.81 (s, 9H, AlMe3), 1.04 (d, 3JH–H = 6.9 Hz, 12H, (CH3)2CH), 1.36 (d, 3JH–H = 6.9 Hz, 12H, (CH3)2CH), 2.85 (sept, 3JH–H = 6.9, 4H, (CH3)2CH), 4.07 (s, 1H, NH), 5.79 (s, 2H, NCHNCH) and 7.07–7.23 (m, 6H, Ar) ppm. 13C NMR (75 MHz, C6D6): δ −6.45, 23.46, 24.58, 29.11, 116.23, 125.19, 130.85, 131.46, 147.46, 152.87 ppm.Preparation of (L-AlMe2)2 (3)
Ligand 1 (0.200 g, 0.496 mmol) and 0.243 mL of a 2.0 M solution of trimethylaluminium in heptane (0.035 g, 0.486 mmol) were combined in a sealed reaction vessel with 10 mL toluene. The resulting mixture was heated to 110 °C overnight where the solvent was then subsequently removed in vacuo (yield: 0.620 g, 73%). Analytically pure crystals were isolated by recrystallization in hot heptane cooled slowly to ambient temperature. Mp 320 °C (decomp.); anal. calc. for C58H84Al2N6: C, 78.16; H, 9.05; N, 9.89%; found: C, 75.35; H, 9.01; N, 8.96%; 1H NMR (C6D6, 300 MHz): δ −1.50 (s, 12H, Al(CH3)2), 1.04 (d, 3JH–H = 6.8 Hz, 24H, (CH3)2CH), 1.46 (d, 3JH–H = 6.8 Hz, 24H, (CH3)2CH), 3.27 (sept, 3JH–H = 6.8 Hz, 8H, (CH3)2CH), 5.78 (s, 4H, NCHCHN), 7.15–7.22 (m, 12H, Ar) ppm. 13C NMR (75 MHz, C6D6): δ −5.99, 23.64, 25.66, 28.69, 117.18, 124.61, 130.24, 135.38, 147.85, 150.66 ppm.Isolation of (L–H)2AlMe2(L) (4)
A sealed reaction vessel fitted with a Teflon stopper containing ligand 1 (0.200 g, 0.496 mmol), 2.0 M AlMe3 in heptane (0.124 mL, 0.248 mmol) and 15 mL toluene was heated to 60 °C for 48 h. The solvent was removed in vacuo leaving a colorless solid. Analysis by 1H NMR showed a mixture of products. However, none of these matched with 1, 2 or 5 and only trace amounts of 3 were detected. Crystallization from hot heptane gave a small amount of 4 suitable for analysis by single crystal X-ray diffraction. Multiple attempts to isolate bulk amounts of 4 by carefully monitoring the above reaction or adding ligand 1 to 3 were unsuccessful.Preparation of L2AlMe (5)
A sealed reaction vessel fitted with a Teflon stopper containing compound 2 (3.00 g, 6.32 mmol) and ligand 1 (2.55 g, 6.32 mmol) in 15 mL toluene was heated to 110 °C for 48 h. The solvent was removed in vacuo and the remaining solid was dissolved in hot heptane, which was slowly cooled to ambient temperature yielding the product as beige crystals (yield: 3.89 g, 72%). Analytically pure crystals were recovered by a second recrystallization in hot heptane cooled slowly to ambient temperature. Mp 226.9–228.4 °C. Anal. calc. for C55H75AlN6: C, 78.16; H, 9.05; N, 9.89%; found: C, 77.99; H, 8.95; N, 9.89%. 1H NMR (300 MHz, C6D6): δ −1.49 (s, 3H, AlCH3), 1.12 (d, 3JH–H = 6.9 Hz, 24H, (CH3)2CH), 1.18 (d, 3JH–H = 6.9 Hz, 24H, (CH3)2CH), 3.11 (sept, 3JH–H = 6.9 Hz, 8H, (CH3)2CH), 5.97 (s, 4H, NCHNCH) and 7.13–7.29 (m, 12H, Ar) ppm. 13C NMR (75 MHz, C6D6): δ −11.10, 23.37, 24.50, 28.74, 112.94, 124.00, 128.80, 135.37, 143.11, 148.09 ppm.Reaction of 5 with triflic acid
(a) In a scintillation vial, triflic acid (0.010 mL, 0.12 mmol) dissolved in 5 mL of diethyl ether was added dropwise to a rapidly stirring solution of 5 (0.100 g, 0.12 mmol) in 5 mL of diethyl ether. A colorless precipitate rapidly formed and gas evolution (methane) was observed. After stirring for 15 minutes, the solvent was decanted from the solids and the solids were washed with diethyl ether. The residual solvent was removed from the solids in vacuo. 1H NMR analysis of the solids revealed loss of the Al–Me signals. In order to partially determine the composition of the solid, a few X-ray quality crystals were grown from a THF solution layered with pentane. The analyzed crystal, compound 6, matched the 1H NMR data.1H NMR (300 MHz, THF-d8): δ 1.23 (d, 3JH–H = 6.9 Hz, 24H, (CH3)2CH), 1.26 (d, 3JH–H = 6.9 Hz, 24H, (CH3)2CH), 1.79 (m, 8H, THF), 2.84 (sept, 3JH–H = 6.9 Hz, 8H, (CH3)2CH), 3.62 (m, 8H, THF), 6.94 (s, 4H, NCHNCH) and 7.33–7.47 (m, 12H, Ar) ppm. Note: protons attached to the imine functionality were not observed, presumably due to their rapid exchange with one another.
(b) In a second attempt, the reaction scale was multiplied 2.5×. Analysis of this material gave a slightly different 1H NMR spectrum than in the smaller scale reaction, but was still not diagnostic. A few X-ray quality crystals were grown from a THF solution layered with pentane. Analysis of these crystals revealed the formation of compound 7. 1H NMR (300 MHz, THF-d8): δ 1.22 (d, 3JH–H = 6.9 Hz, 12H, (CH3)2CH), 1.25 (d, 3JH–H = 6.9 Hz, 12H, (CH3)2CH), 2.74 (sept, 3JH–H = 6.9 Hz, 4H, (CH3)2CH), 7.02 (s, 2H, NCHNCH) and 7.33–7.45 (m, 6H, Ar) ppm. Note: the two protons attached to the imine functionality were not observed, presumably due to their rapid exchange between the imine and triflate anion.
X-ray crystallography
Crystals of compounds 2–7 were mounted from Paratone-N oil on an appropriately sized MiTeGen MicroMount. The data were collected on a Bruker APEX II charge-coupled-device (CCD) diffractometer, with an Oxford 700 Cryocool sample cooling device. The instrument was equipped with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å; 30 mA, 50 mV), with MonoCap X-ray source optics. For data collection, four ω-scan frame series were collected with 0.5° wide scans, 60 second frames and 416 frames per series at varying ϕ angles (ϕ = 0°, 90°, 180°, 270°). Data collection, unit cell refinement, data processing and multi-scan absorption correction were applied using the APEX II software package.61 The structures were solved using direct methods62 and all non-hydrogen atoms were refined anisotropically using the shelXle63 graphical user interface and the SHELX suite of programs.62 Unless otherwise noted, all hydrogen atom positions were idealized and rode on the atom to which they were attached. The final refinement included anisotropic temperature factors on all non-hydrogen atoms. Details of crystal data, data collection, and structure refinement are listed in Table 1. All figures were made using ORTEP-3 for Windows.64 Additional details of the data collection and structure refinement and tables of bond lengths and angles are given in the ESI. CCDC 1479384–1479389 contain the supplementary crystallographic data for complexes 2–7.†| Compound | 2 | 3 | 4 | 5 | 6 | 7 |
| Chemical formula | C67H100Al2N6 | C56H79AlN6 | C58H84Al2N6 | C55H75AlN6 | C66H91AlF12N6O14S4 | C28H38F3N3O3S |
| Formula mass | 1043.48 | 863.23 | 919.27 | 847.19 | 1575.66 | 553.67 |
| Crystal system | Triclinic | Monoclinic | Triclinic | Monoclinic | Monoclinic | Orthorhombic |
| a/Å | 9.3542(18) | 12.9094(10) | 12.064(2) | 22.0069(19) | 12.6935(15) | 10.5014(8) |
| b/Å | 9.5893(18) | 18.6830(14) | 22.957(4) | 11.7183(10) | 16.0668(19) | 14.0346(10) |
| c/Å | 19.621(4) | 21.6167(16) | 24.196(4) | 20.0358(17) | 19.437(2) | 20.5119(15) |
| α/° | 90.186(3) | 90 | 62.143(2) | 90 | 90 | 90 |
| β/° | 96.123(2) | 90.0500(10) | 75.811(2) | 99.4460(10) | 102.1980(10) | 90 |
| γ/° | 111.258(2) | 90 | 89.463(2) | 90 | 90 | 90 |
| Unit cell volume/Å3 | 1629.2(5) | 5213.7(7) | 5700.7(17) | 5096.8(8) | 3874.6(8) | 3023.1(4) |
| Temperature/K | 125(2) | 125(2) | 125(2) | 125(2) | 125(2) | 125(2) |
| Space group | P |
P21/n | P |
P21/c | P21 | P212121 |
| Z | 1 | 4 | 4 | 4 | 2 | 4 |
| Absorption coefficient, μ/mm−1 | 0.086 | 0.080 | 0.091 | 0.081 | 0.225 | 0.157 |
| No. of reflections measured | 10772 |
50368 |
55016 |
32942 |
27730 |
20332 |
| No. of independent reflections | 5605 | 9160 | 19990 |
8897 | 14581 |
5314 |
| Rint | 0.0275 | 0.0893 | 0.0496 | 0.0290 | 0.0256 | 0.0282 |
| Final R1 values (I > 2σ(I)) | 0.0536 | 0.0490 | 0.0641 | 0.0418 | 0.0456 | 0.0343 |
| Final wR(F2) values (I > 2σ(I)) | 0.1424 | 0.0998 | 0.1718 | 0.0996 | 0.1060 | 0.0763 |
| Final R1 values (all data) | 0.0697 | 0.0945 | 0.0917 | 0.0566 | 0.0579 | 0.0399 |
| Final wR(F2) values (all data) | 0.1544 | 0.1207 | 0.1927 | 0.1083 | 0.1134 | 0.0798 |
| Goodness of fit on F2 | 1.148 | 1.009 | 1.112 | 1.024 | 1.022 | 1.032 |
| CCDC number | 1479384 | 1479388 | 1479389 | 1479386 | 1479385 | 1479387 |
Acknowledgements
We thank the Natural Sciences and Engineering Research Council of Canada (through the Discovery Grants Program to JDM). JDM acknowledges support from the Canadian Foundation for Innovation and the Nova Scotia Research and Innovation Trust Fund. Dr Katherine Robertson is thanked for useful crystallographic advice.References
- L. Yan, X. Wang and M. Zhou, Inorg. Chem. Commun., 2016, 65, 32–34 CrossRef CAS.
- M. Zhou, Q. Yang, H. Tong, L. Yan and X. Wang, RSC Adv., 2015, 5, 105292–105298 RSC.
- M. Zhou, H. Tong, X. Wei and D. Liu, J. Organomet. Chem., 2007, 692, 5195–5202 CrossRef CAS.
- R. Berlinck, A. Burtoloso, A. Trindade-Silva, S. Romminger, R. Morias, K. Bandeira and C. Mizuno, Nat. Prod. Rep., 2010, 27, 1871–1907 RSC.
- R. Berlinck, Nat. Prod. Rep., 2002, 19, 617–649 RSC.
- L. Heys, C. Moore and P. Murphy, Chem. Soc. Rev., 2000, 29, 57–67 RSC.
- F. Edelmann, Chem. Soc. Rev., 2009, 38, 2253–2268 RSC.
- G. R. Giesbrecht, G. D. Whitener and J. Arnold, J. Chem. Soc., Dalton Trans., 2001, 923–927 RSC.
- S. M. Mullins, A. P. Duncan, R. G. Bergman and J. Arnold, Inorg. Chem., 2001, 40, 6952–6963 CrossRef CAS PubMed.
- I. Karmel, N. Fridman, M. Tamm and M. S. Eisen, Organometallics, 2015, 34, 2933–2942 CrossRef CAS.
- I. Karmel, M. Botoshansky, M. Tamm and M. S. Eisen, Inorg. Chem., 2014, 53, 694–696 CrossRef CAS PubMed.
- A. Milanov, R. Fischer and A. Devi, Inorg. Chem., 2008, 47, 11405–11416 CrossRef CAS PubMed.
- W. P. Kretschmer, C. Dijkhuis, A. Meetsma, B. Hessen and J. H. Teuben, Chem. Commun., 2002, 608–609 RSC.
- N. Kuhn, R. Fawzi, M. Steimann, J. Wiethoff, D. Bläser and R. Boese, Z. Naturforsch., 1995, 50b, 1779–1784 Search PubMed.
- A. G. Trambitas, T. K. Panda and M. Tamm, Z. Anorg. Allg. Chem., 2010, 636, 2156–2171 CrossRef CAS.
- M. Tamm, D. Petrovic, S. Randoll, S. Beer, T. Bannenberg, P. G. Jones and J. Grunenberg, Org. Biomol. Chem., 2007, 5, 523–530 CAS.
- K. Naktode, S. Das, J. Bhattacharjee, H. Nayek and T. Panda, Inorg. Chem., 2016, 55, 1142–1153 CrossRef CAS PubMed.
- K. Nomura, S. Patamma, H. Matsuda, S. Katao, K. Tsutsumi and H. Fukuda, RSC Adv., 2015, 5, 64503–64513 RSC.
- W. Apisuk, A. G. Trambitas, B. Kitiyanan, M. Tamm and K. Nomura, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2575–2580 CrossRef CAS.
- D. Shoken, M. Sharma, M. Botoshansky, M. Tamm and M. S. Eisen, J. Am. Chem. Soc., 2013, 135, 12592–12595 CrossRef CAS PubMed.
- M. Sharma, H. Yameen, B. Tumanskii, S. Filimon, M. Tamm and M. S. Eisen, J. Am. Chem. Soc., 2012, 134, 17234–17244 CrossRef CAS PubMed.
- S. H. Stelzig, M. Tamm and R. M. Waymouth, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6064–6070 CrossRef CAS.
- M. Tamm, S. Randoll, E. Herdtweck, N. Kleigrewe, G. Kehr, G. Erker and B. Rieger, Dalton Trans., 2006, 21, 459–467 RSC.
- M. Tamm, S. Randoll, T. Bannenberg and E. Herdtweck, Chem. Commun., 2004, 876–877 RSC.
- N. Kuhn, R. Fawzi, M. Steimann and J. Wiethoff, Z. Anorg. Allg. Chem., 1997, 623, 769–774 CrossRef CAS.
- A. Trambitas, D. Melcher, L. Hartenstein, P. Roesky, C. Daniliuc, P. Jones and M. Tamm, Inorg. Chem., 2012, 51, 6753–6761 CrossRef CAS PubMed.
- N. Kuhn, M. Göhner, M. Grathwohl, J. Wiethoff, G. Frenking and Y. Chen, Z. Anorg. Allg. Chem., 2003, 629, 793–802 CrossRef CAS.
- D. W. Stephan, Organometallics, 2005, 24, 2548–2560 CrossRef CAS.
- K. Dehnicke and A. Greiner, Angew. Chem., Int. Ed. Engl., 2003, 42, 1340–1354 CrossRef CAS PubMed.
- K. Dehnicke, M. Krieger and W. Massa, Coord. Chem. Rev., 1999, 182, 19–65 CrossRef.
- I. Karmel, N. Fridman, M. Tamm and M. S. Eisen, J. Am. Chem. Soc., 2014, 136, 17180–17192 CrossRef CAS PubMed.
- A. Trambitas, T. Panda, J. Jenter, P. Roesky, C. Daniliuc, C. Hrib, P. Jones and M. Tamm, Inorg. Chem., 2010, 49, 2435–2446 CrossRef CAS PubMed.
- J. Volbeda, P. G. Jones and M. Tamm, Inorg. Chim. Acta, 2014, 422, 158–166 CrossRef CAS.
- T. Glöge, D. Petrovic, C. Hrib, P. G. Jones and M. Tamm, Eur. J. Inorg. Chem., 2009, 4538–4546 CrossRef.
- D. Petrovic, T. Glöge, T. Bannenberg, C. G. Hrib, S. Randoll, P. G. Jones and M. Tamm, Eur. J. Inorg. Chem., 2007, 3472–3475 CrossRef CAS.
- D. Petrovic, T. Bannenberg, S. Randoll, P. G. Jones and M. Tamm, Dalton Trans., 2007, 2812–2822 RSC.
- M. Đurović, J. Bogojeski, B. Petrović, D. Petrović, F. W. Heinemann and Ž. D. Bugarčić, Polyhedron, 2012, 41, 70–76 CrossRef.
- J. Bogojeski, R. Jelic, D. Petrovic, E. Herdtweck, P. G. Jones, M. Tamm and Z. D. Bugarcic, Dalton Trans., 2011, 40, 6515–6523 RSC.
- R. Kinjo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 5930–5933 CrossRef CAS PubMed.
- O. Back, B. Donnadieu, M. von Hopffgarten, S. Klein, R. Tonner, G. Frenking and G. Bertrand, Chem. Sci., 2011, 2, 858–861 RSC.
- F. Dielmann, O. Back, M. Henry-Ellinger, P. Jerabek, G. Frenking and G. Bertrand, Science, 2012, 337, 1526–1528 CrossRef CAS PubMed.
- F. Dielmann, C. E. Moore, A. L. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2013, 135, 14071–14073 CrossRef CAS PubMed.
- F. Dielmann, D. M. Andrada, G. Frenking and G. Bertrand, J. Am. Chem. Soc., 2014, 136, 3800–3802 CrossRef CAS PubMed.
- T. Ochiai and S. Inoue, Phosphorus, Sulfur Silicon Relat. Elem., 2016, 191, 624–627 CrossRef CAS.
- T. Ochiai, D. Franz, X. Wu and S. Inoue, Dalton Trans., 2015, 44, 10952–10956 RSC.
- D. Franz, T. Szilvási, E. Irran and S. Inoue, Nat. Commun., 2015, 6, 10037–10042 CrossRef CAS PubMed.
- D. Franz, E. Irran and S. Inoue, Dalton Trans., 2014, 43, 4451–4461 RSC.
- D. Franz and S. Inoue, Chem.–Eur. J., 2014, 20, 10645–10649 CrossRef CAS PubMed.
- M. W. Lui, N. R. Paisley, R. McDonald, M. J. Ferguson and E. Rivard, Chem.–Eur. J., 2016, 22, 2134–2145 CrossRef CAS PubMed.
- M. W. Lui, C. Merten, M. J. Ferguson, R. McDonald, Y. Xu and E. Rivard, Inorg. Chem., 2015, 54, 2040–2049 CrossRef CAS PubMed.
- N. A. Giffin, M. Makramalla, A. D. Hendsbee, K. N. Robertson, C. Sherren, C. C. Pye, J. D. Masuda and J. A. C. Clyburne, Org. Biomol. Chem., 2011, 9, 3672–3680 CAS.
- N. A. Giffin and J. D. Masuda, Coord. Chem. Rev., 2011, 255, 1342–1359 CrossRef CAS.
- O. Puntigam, D. Förster, N. A. Giffin, S. Burck, J. Bender, F. Ehret, A. D. Hendsbee, M. Nieger, J. D. Masuda and D. Gudat, Eur. J. Inorg. Chem., 2013, 2041–2050 CrossRef CAS.
- N. A. Giffin, A. D. Hendsbee, T. L. Roemmele, M. D. Lumsden, C. C. Pye and J. D. Masuda, Inorg. Chem., 2012, 51, 11837–11850 CrossRef CAS PubMed.
- A. D. Hendsbee, N. A. Giffin, Y. Zhang, C. C. Pye and J. D. Masuda, Angew. Chem., Int. Ed., 2012, 51, 10836–10840 CrossRef CAS PubMed.
- C. M. Ong, P. McKarns and D. W. Stephan, Organometallics, 1999, 18, 4197–4204 CrossRef CAS.
- J. Müller, U. Ruschewitz, O. Indris, H. Hartwig and W. Stahl, J. Am. Chem. Soc., 1999, 121, 4647–4652 CrossRef.
- T. K. Panda, A. G. Trambitas, T. Bannenberg, C. G. Hrib, S. Randoll, P. G. Jones and M. Tamm, Inorg. Chem., 2009, 48, 5462–5472 CrossRef CAS PubMed.
- C. Cui, H. W. Roesky, H. Schmidt, M. Noltemeyer, H. Hao and F. Cimpoesu, Angew. Chem., Int. Ed., 2000, 39, 4274–4276 CrossRef CAS.
- T. Mandai, H. Masu and P. Johansson, Dalton Trans., 2015, 44, 11259–11263 RSC.
- APEX2 Ver. 2008.5,Bruker AXS, Inc., Madison, Wisconsin, USA, 2008 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Selected NMR spectra and crystallographic data can be found. CCDC 1479384–1479389. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra15507c |
| This journal is © The Royal Society of Chemistry 2016 |