
TiO2 sol-embedded in electroless Ni–P coating: a novel approach for an ultra-sensitive sorbitol sensor†
Abstract
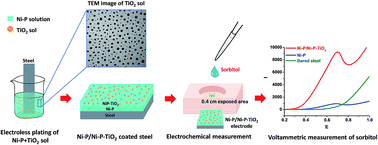
A Ni–P–TiO2 coating was readily prepared by direct incorporation of nano-TiO2 sol into a Ni–P solution followed by electroless deposition. This coating was applied as a working electrode in an electrochemical sensor for the first time. The morphologies of the TiO2 sol and the coated surface were well characterized by TEM, SEM and AFM. The high hydrophilicity of this surface was verified by contact angles of 40.7/41.8. Here, the appropriate amount of TiO2 within the nanocomposite was optimized (2 g L−1) prior to applying as an electrode. Interestingly, the electrocatalytic activity of the coating towards the oxidation of alcoholic compounds was investigated by linear sweep voltammetry. Apparently, incorporation of TiO2 into the composites substantially improved the electrocatalytic activity of Ni–P and 2 layers of Ni–P/Ni–P–TiO2 coating provided the highest sensitivity for all analytes, especially for sorbitol. A low LOD value of 1.0 nM and a wide linear range of 2.0 nM to 0.2 mM were achieved for sorbitol. Furthermore, a high stability and high reproducibility (2.96% RSD) for this system were obtained. Owing to ultra-high sensitivity, wide linearity, high stability, easy preparation and low cost, it might be a promising tool for early diagnosis of diabetes via sorbitol detection.
 Please wait while we load your content...
Please wait while we load your content...

