
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Upcycling spent petroleum cracking catalyst: pulsed laser deposition of single-wall carbon nanotubes and silica nanowires
N. Souzaa,
F. Lasserrea,
A. Blickleyab,
M. Zeigerac,
S. Suáreza,
M. Duarted,
V. Presserac and
F. Mücklich*a
aDepartment of Materials Science, Saarland University, 66123 Saarbrücken, Germany. E-mail: muecke@matsci.uni-sb.de
bDepartment of Materials Science and Engineering, Drexel University, Philadelphia, PA 10104, USA
cINM – Leibniz Institute for New Materials, 66123 Saarbrücken, Germany
dEngineering and Technology School, Catholic University of Uruguay, 11600 Montevideo, Uruguay
First published on 25th July 2016
Abstract
Fluid catalytic cracking (FCC), which currently accounts for half of the worldwide petroleum refining efforts, relies on catalytic, aluminosilicate zeolite particles which slowly deactivate. As of yet, this FCC catalyst residue (FC3R) has no commercial outlet, resulting in abundant amounts of landfill-destined refuse. However, this overlooked waste has the right ingredients for the synthesis of some of today's emerging nanomaterials. High-carbon FC3R, sourced from a Uruguayan refinery, was identified as faujasite particles encased in graphitic carbon shells. We show that pulsed laser ablation of raw FC3R produces simultaneous deposition of single-wall carbon nanotubes and silica nanowires through vapour/solid–liquid–solid self-assembly in distinct zones of an oven-laser apparatus. This is an extreme revalorisation and provides a new untapped resource for research and applications in C- and Si-based nanomaterials and mesoscopic physics.
1. Introduction
Petroleum refining relies on cracking of large gasoil molecules into smaller vapour-phase distillable products. Depending on geographical location, specific fuel needs and crude origin, refineries employ different catalytic cracking installations. Fluid catalytic cracking (FCC) and hydroprocessing (-treating & -cracking) (HP) comprise 48% and 24% of the installed capacities in refineries world-wide.1 FCC occurs at high temperatures with the help of a fluidised catalyst powder, modern versions of which consist mainly of faujasite, as well as an amorphous alumina matrix, a silica sol binder and a kaolinite clay (silica and alumina) filler.1 The zeolite faujasite (FAU) is a crystalline aluminosilicate mineral (2–5 Si/Al atomic ratio) with a primitive cell consisting of 18-membered sodalite cages, orthogonally connected by hexagonal prisms. These result in a primary supercage and secondary pore structures which confine the hydrocarbons and, together with coordinated oxygen and sodium ions (192 [(Si/Al)O4]4−/5− tetrahedrons per unit cell), act as Brönsted/Lewis acids and crack them.2 Of the 200-some documented types of zeolite, two types of FAU exist, X and Y, each in a variety of modifications (de-/sodiated, de-/hydrated, ultrastable, very ultrastable, dealuminated3). Although zeolite Y is the current FCC standard, its composition (modification) can also vary to adapt to real-time crude input and market output needs.1During cracking, petroleum coke (PC) accumulates on different locations and components of the FCC unit (conduits, chambers, chimneys). This form of coke, specific to the petrochemical industry, is a high carbon content fuel, also rich in sulphur and metals, that infiltrates (i.e., poisons) and deposits on (i.e., fouls) the catalyst, significantly reducing the catalytic activity.4 The PC is thus periodically burned off to maintain activity in a regenerator, the produced heat is refed into the system (FCC C–C cleaving is endothermic) and the catalyst is recirculated for further catalysis. A fraction of this circulating equilibrium catalyst (ECAT) is thus continually renewed in order to maintain activity and reduce metal and sulphur contents.5 Over time, however, the catalyst suffers stronger yield decline with irreversible decay due to poisoning and fouling (PC, metals and sulphur) and sintering (collapse of crystalline structure and loss of surface area),4 requiring further cyclone removal or electrostatic precipitation, yielding the eponymous electrostatic precipitator catalyst (EPCAT).6 Thus, ECAT & EPCAT are extracted in the form of off-white to black powders/rocks, depending on the coke content, referred to as FCC catalyst residue (FC3R), or in a broader sense, as catalyst coke or catalyst fines (catfines), to include hydroprocessing catalyst residue (HPCR). At this stage, these residues can be rejuvenated for reuse, a limited amount of times,7 eventually becoming waste. For the purpose of this work, spent catalyst or FC3R will designate that which is non-regenerable with no residual functional activity.
FC3R is often assigned to PC in yield statistics as a subcategory.8 Although a by-product, upon extraction and/or purification, most PC is marketed as a commonplace global commodity. Disposal is thus avoided, recycling added-value materials back into the industry. Established buyers include power generation, cement, metallurgy and industrial. Indeed, coke (PC or other) can substitute coal for domestic heating purposes and is used in the metallurgical industry as fuel or reducing agent.9 NASA has used coke, combined with other materials, as heat shielding in various missions.10 Yet, marketable coke must be relatively pure carbon; that is why FC3R, with its high amounts of zeolite, is not considered as such. FC3R is the largest catalyst waste category in refineries.11 In 2006, 0.5 MT of FCC catalyst were being used worldwide,12 which will inevitably end up in landfills due to lacking technology, profitability and regulation.
Research is currently exploring outlets for spent cracking catalysts. To this end, in addition to the variety of zeolites (and their modifications) used in petroleum refining, an important distinction arises between FCC and HP catalyst waste, and therefore, their ensuing disposal/recycling. HP catalysts contain very high amounts of metal (Mo, Ni, V, Co, W) and their disposal/reuse implies potential metal leaching. Because of this, although both FC3R and HPCR must undergo treatment in order to meet ever stringent environmental standards, the latter metal-laden catalyst awaits greater hurdles.7 In fact, HPCR is labelled hazardous (US & EU)13,14 and cannot be exported to/from third world countries according to the Basel Convention and OECD rules.15 FC3R, on the other hand, is non-hazardous.14
A European Commission initiative from 1991 foresaw construction materials as a potential outlet for FC3R16 but it was not until 1997 that it was studied.17 Largely dominated by FC3R, spent catalysts have since been tested as substitute or aggregate in a variety of admixtures, mainly for construction. In addition to refractories18,19 and ceramic frits,20 their pozzolanic activity seems to hold the greatest promise: high-performance Portland cements, pastes, binders, mortars and concretes have been successfully manufactured with cement substitution rates of up to 40 wt%.21 The characteristics of these composite products versus their standard counterparts range from similar quality to 117% pozzolanic activity,21 147% compressive strength22 and improved hydraulics, durability, bleeding and setting times. Despite these very promising results, spent catalysts are not yet marketed.
In our experiments, the carbon coke in FC3R was considered to provide an interesting alternative carbon feedstock for carbon nanotube (CNT) synthesis. Apart from the necessary carbon, some of the elements present in FC3R are known catalysts in CNT growth. Although Al and Si do not directly take part in CNT growth,23 their oxides have been extensively used as growth support24 and Si can enhance growth through oxidation.25 The 48% void fraction of FAU can be conducive to higher CNT yields.26 Furthermore, trapped oxygen could burn off amorphous carbon, increasing CNT purity. Finally, sulphur is known to broaden the diameter distribution27 and to produce Y-junctions through structural defects.28
In addition, the aluminosilicate zeolite content in FC3R was expected to be conducive to silicon-based nanowires, namely silica nanowires (SiONWs). These lesser known nano-homologues exhibit excellent (blue) photoluminescence, which has dominated their study as well as their applications, dating back to their discovery in 1998.29 Together with gold nanoparticles they feature a surface plasmon resonance absorption that can be used for photothermal cancer therapy.30 Bolstered by their physical and chemical stability, large surface area, and biocompatibility, SiONWs are also envisioned and studied in biosensing31 and solar cells.32 Their chemistry is easily tuneable thanks to well-established protocols for surface modification.33,34 These rods have an ultimate strength one and two orders of magnitude above that of Kevlar and high-strength steel, respectively.35
Pulsed laser deposition (PLD),36 a branch of laser ablation (LA), was envisaged as a suitable means for these experiments. PLD is one of the principal established techniques for the growth of nanowires and -tubes. Although continuous wave (CW) lasers can ablate without the help of an oven,37 pulsed lasers are usually preferred for LA due to the higher peak power output, accumulated between each pulse and released in very short, discrete ns pulses.38 This renders PLD more controllable with respect to the plume dynamics emanating from the solid feedstock (target).38 Chemical vapour deposition (CVD), another standard technique for either nanostructure, is generally fed by a gaseous precursor. Solids such as FC3R would require costly and laborious liquid-state chemistry to produce sprayable or vaporisable solutions/suspensions, easily surpassing the cost of the synthesis itself. Thermal evaporation would not manage the variety of species present in the FC3R. In arc discharge (Arc), a more energy-intensive CNT synthesis route, the anode is consumed in favour of CNT-containing soot that builds up on the cathode. Compared to the required standard, high-purity graphite electrodes, the aluminosilicates in the FC3R would not guarantee the necessary conductivity. Supposing it would, its anisotropy and alternating compositions of layered cokes, would alter the field lines and cause the arc to wander, rendering the discharge plasma unstable and unfavourable to prolonged nanotube growth. Not to mention, Arc is incompatible with SiONW growth and the aluminosilicates would be lost in the process. Finally, contrary to ablated graphite, which contributes single atoms to the CNTs, larger aromatic species present in the coke can be directly incorporated by PLD.39
Common precursors for CNTs are carbon-containing gases and graphite. Si sources for SiONW precursors include: silicon wafers, silane, silicon-based powders (Si, SiC, SiO2) and silica xerogel (mesoporous silica); oxygen can be found in the Si sources, metallic oxides can be fed in the gas phase or in impurities from other process gases (e.g., H2, N2, Ar), and/or mixtures thereof. Catalysts for either nanostructure are usually metals, for example, Fe, Co, Ni, Ga, Au, Sn or Pd.40,41 A number of substances have been used to activate, enhance growth or produce catalyst, such as activated carbon, graphite and metallic nitrates.
Given their popularity and impending market boom, CNT synthesis has experimented with a manifold of alternative carbon sources amongst vegetable (camphor,42–48 turpentine oil,49,50 eucalyptus oil,51 palm oil52), mineral (coal and coal gas53–68), nano- (C60 fullerenes,69–71 CNTs72–76) and petrochemical carbons (heavy oil residue,77 tire powder,78 carbon fibre,79 carbon black,80,81 asphalt82,83). With the exception of tire powder, while some might provide cheaper alternatives, none of them represent waste, having established markets and uses. The comparatively understudied SiONWs have not required alternative precursors from recycling or waste and are yet to be explored.
Two recent reports exist on the use of marketable (zeolite-free) PC for CNT synthesis. Kai Xu et al. were able to controllably produce single-wall (SW) and double-wall CNTs by arc discharge of an Fe–PC electrode in He and Ar atmospheres, respectively.84 Crushed, sieved coke and Fe powders were mixed at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 weight ratio and filled into a hollowed out high-purity graphite anode. The high-purity graphite cathode was not the deposit collector in this case, but rather a metal wire net placed near the electrodes. The high-quality tubes presented narrow diameter distributions, more so for the SWCNTs. Abdullayeva et al. found that a coke rock, gasified at intermediate temperature zones in a tube furnace under He and H2, produced CNTs on FeCl2 thin films and powders, and Fe thin films.85
:2 weight ratio and filled into a hollowed out high-purity graphite anode. The high-purity graphite cathode was not the deposit collector in this case, but rather a metal wire net placed near the electrodes. The high-quality tubes presented narrow diameter distributions, more so for the SWCNTs. Abdullayeva et al. found that a coke rock, gasified at intermediate temperature zones in a tube furnace under He and H2, produced CNTs on FeCl2 thin films and powders, and Fe thin films.85
FAU is not new to SiONW and CNT synthesis either, having served as a growth template for pore-controlled diameter distributions or catalyst arrangements. Reports of thermal disproportionation of SiO show the porous structure of FAU (cages and channels) defining the core diameter of resulting core/shell hybrid nanowires. Primarily, fine, crystalline Si cores sheathed with amorphous SiO2 were found protruding from the zeolite pores. Beyond the templated growth, the FAU provided the silica in the shells. To a lesser extent, inverted (silica core) and side-by-side (half wire) nanowires were found at undisclosed locations in the sample.86,87 In CNT synthesis, zeolites can similarly provide catalytic decomposition,88 catalyst support89 and structural templates.90 In fact, the thinnest free-standing SWCNTs (0.4 nm) were grown inside the channels of AFI zeolite (AlPO45−).90
Given these existing synergies, the intention of this study is to provide a proof of concept for the feasibility of CNT and SiONW synthesis from FC3R by PLD, including intermittent parallels to cracking catalyst waste in general. The motivation is the revalorisation of this otherwise wasted material, addressing synthesis cost efficiency with cheaper precursors, beyond economies of scale and process optimisation. A FC3R rock from the state-owned La Teja refinery in Uruguay (operated by ANCAP), that in 2004 was producing 65 T of cracking catalyst waste a year,91 was thus used as the starting material for this work, constituting the carbon and silica precursor for CNT and SiONW synthesis by PLD. The produced nanostructures are technologically relevant 0-, 1- and 2-D nanomaterials: the expected SiONWs and SWCNTs, as well as onion-like carbons (OLCs), carbon nanodots (CNDs) and SiC nanostructures. Feasibility and proof are provided by a thorough morphological and chemical characterisation of the precursor (FC3R in 3.1) and resulting nanostructures (SiONWs in 3.2; SWCNTs in 3.3), supported by a combination of electron microscopy, micromilling and spectroscopic techniques.
2. Experimental
2.1 Thin film deposition
ca. 100 nm thin Ni films were DC magnetron-sputtered onto commercial SiO/Si wafers (500 ± 25 μm Si + 250 nm SiO2) by physical vapour deposition (PVD). A base chamber pressure of 3.0 × 10−5 bar, an Ar pressure of 4.9 × 10−3 bar and a power of 103 W produced a sputtering rate of 14 nm min−1.2.2 Nanomaterial synthesis
Samples were synthesised by PLD. The carbon feedstock pellets (targets) were obtained by grinding a FC3R rock, faceting it into a manageable shape, appropriate for the PLD apparatus. The target was vaporised by a pulsed laser (solid state Nd:YAG Newport-SpectraPhysics Quanta-Ray 290) inside a tube furnace (Fig. 1) with the parameters in Table 1 and a total radiation time of 30 min.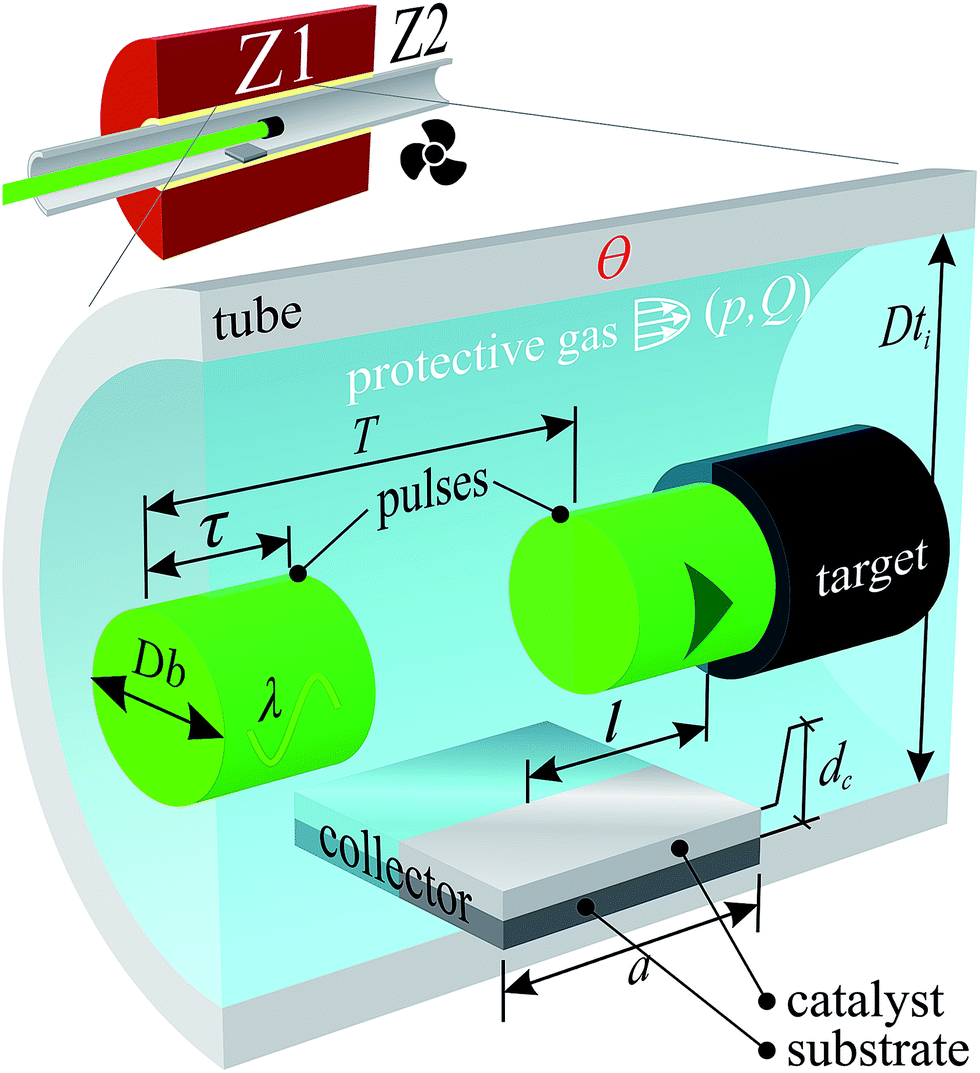 | ||
| Fig. 1 Zone Z1 of PLD oven schematics (inset): tube (interior diameter Dti) furnace flooded with protective gas (pressure p, flow Q) at temperature Ө with incoming laser pulses (wavelength λ, pulse duration τ, frequency 1/T, beam diameter Db) impinging on a carbonaceous target and depositing on a square collector (thin-film catalyst (unrelated to cracking catalyst) (thickness dc) on SiO2/Si substrate (side a)) at a distance l from the laser impingement. Zone Z2 is the downstream, cooled tube furnace exit. | ||
| Chamber | Quartz tube | Dti = 29 mm | Ө = 1200 °C | |||
| Atmosphere | Ar | Purity 6.0 | p = 1.3 bar | Q = 3 sccm | ||
| Radiation | λ = 532 nm | τ = 10 ns | 1/T = 10 Hz | Db = 8 mm | F = 1.59 J cm−2 | Vertical polarisation |
| Target | Faceted FC3R rock | Composition: see results | ||||
| Collector | Catalyst: Ni | dc = 100 nm | Substrate: SiO2/Si | l ≈ 30 mm | a = 10 mm | |
2.3 Material characterisation
Scanning electron microscopy (SEM) (at 10–15 kV), focused ion beam (FIB) (Ga+ at 30 kV) milling and energy-dispersive X-ray spectroscopy (EDS) (10–20 kV) were carried out in a dual-beam workstation (FEI Helios NanoLab 600) equipped with an EDS detector (EDAX Company). FIB cross-sections require the deposition of a Pt layer directly above the revealed cross-section to avoid curtaining effects.Transmission electron microscopy (TEM; JEOL 2100F) was carried out at 200 kV on drop-casted dispersions in ethanol on a copper grid with a lacey carbon film (Gatan).
Raman spectra from 100 to 3500 cm−1 were obtained in a Raman spectrometer (Renishaw inVia) with 532 nm (2.33 eV) CW excitation through a 50× objective with a numerical aperture of 0.75, producing a 2.91 μm spot (calculated in ref. 92) on the sample. The power was set to 8.8 μW (0.13 kW m−2), below a threshold of 2.65 kW cm−2, beyond which modification was observed in SWCNTs.92 All Raman spectra were baseline-corrected with a straight line through the middle of the base noise over the complete range, if not specified otherwise, and peaks were fitted with Lorentzian functions.
X-ray diffraction (XRD) was performed (X'Pert MPD) with Cu Kα radiation from a Cu source in a θ–θ geometry configuration. Spectra were acquired from 5–80° 2θ with a step size of 0.026°.
2.4 Decoking
Decoked FC3R was obtained by grinding a sample into fine powder, then heating to 750 °C in air and mixing, thus thermally oxidising and removing the organic and volatile coke species without thermally decomposing the zeolite (at 793 °C).933. Results and discussion
3.1 FC3R
The obtained raw FC3R rocks are agglomerates of coke-covered (shell) catalyst particles (core) which present an average core diameter and shell thickness of ca. 40 μm (Fig. 2a) and 2.65 μm (inset Fig. 2a). This would imply 69 vol% and 66 wt% catalyst (dFAU = 1.93 g cm−3;94 dgraphite = 2.16 g cm−3 (ref. 95)). However, decoking revealed 57 wt% catalyst, indicating excess interparticle carbon.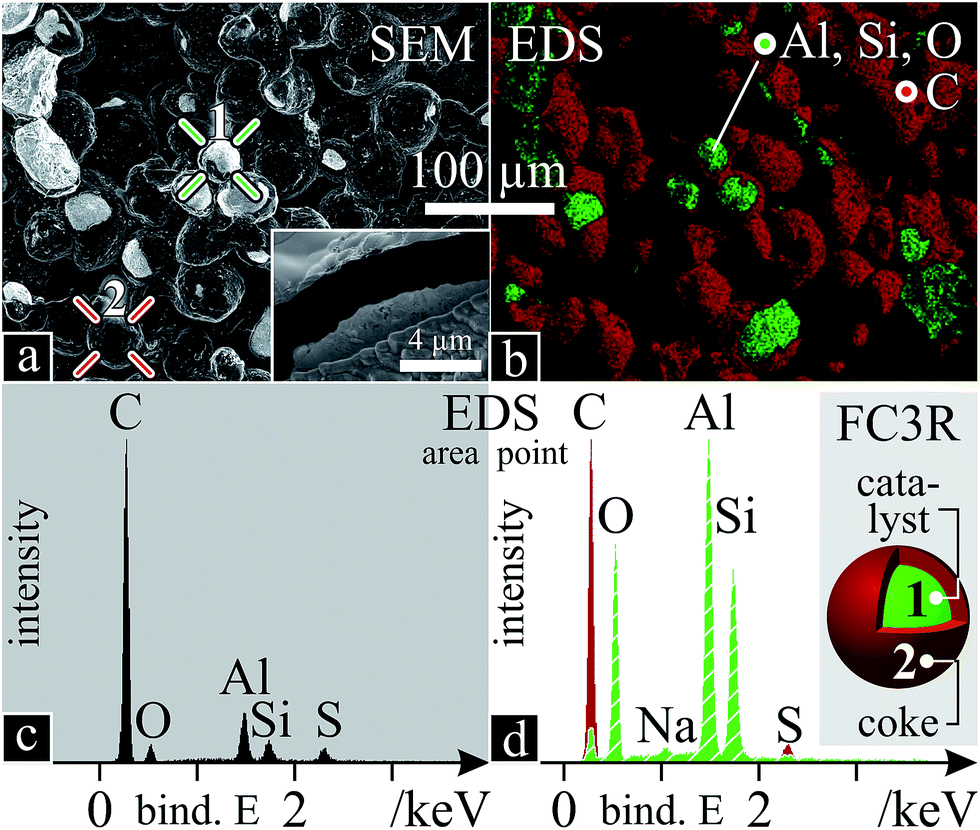 | ||
| Fig. 2 FC3R morphology and chemistry: (a) fracture SEM micrograph with points 1 (catalyst core; green crosshair) and 2 (coke shell; red crosshair) and close-up of FIB cross-section (inset); (b) EDS colour map of Al, Si, O and C; (c) EDS scan of area in “a”; (d) EDS spectra of points 1 (green, striped) and 2 (red) in “a”, with a schema of a FC3R particle and what points 1 and 2 correspond to. S was equally present in both cores and shells of the particles. All EDS data refer to K-shell electrons. The dark patches in “b” are out of plain. | ||
An elemental EDS analysis reveals C, Al, Si, O and traces of S and Na. Colour-mapped scans for each element (Fig. 2b) assign the C to the coke shells and Al, Si, O to the catalyst. A global spectrum of the area shows predominant C (Fig. 2c) due to a majority of shells and EDS penetration depth (2 μm, at 10 kV, below shell thickness), however local spectra of the coke and the catalyst show distinct species (Fig. 2d), with Na attributed to the latter. Na ions are used to balance the alumina tetrahedra in the zeolite during its synthesis.96
Beyond elemental analysis, XRD of the FC3R produced unintelligible signal convolution due to its complex composition. Isolation of the carbonaceous species was not possible, yet, it is expected to contain mainly aromatic carbon, namely, two to five 5/6-membered aromatic carbon rings, from C12H10 to C18H12.85 On the other hand, X-ray powder diffraction of the isolated catalyst (decoked FC3R) (Fig. 3b) was successful, and contrasted with a selection of simulated zeolite XRD powder patterns3,97 and experimental alumina98 and silica99 patterns. Two groups of distinct reflections, narrow and well-defined vs. broad, clearly evidence the convoluted signal of two discrete species in the FC3R, unequivocally identified as sodiated zeolite Y (FAU NaY: |Na28.8Ca14.4(H2O)263|[Si134.4Al57.6O384])3 (Fig. 3a) and gamma alumina (γ-Al2O3) (Fig. 3c, right), respectively. The latter is present as a baseline to the former, an overlay of both. The Scherrer equation,100 based on these peak geometries, qualitatively attributes much smaller crystallites to the alumina in contrast to the longer-range order of FAU. We can conclude that this alumina polymorph is not that of the FAU aluminate tetrahedra (AlO4) and likely stems from the kaolinite or the matrix, components of fluid catalytic cracking (FCC) catalysts. Silica could only be marginally attributed to four unidentified peaks in the FC3R (Fig. 3c, left). Nonetheless, the presumed FAU and aluminosilicates could be confirmed with sufficient detail and certainty for the framework of this study, indicating an industry standard FCC catalyst.
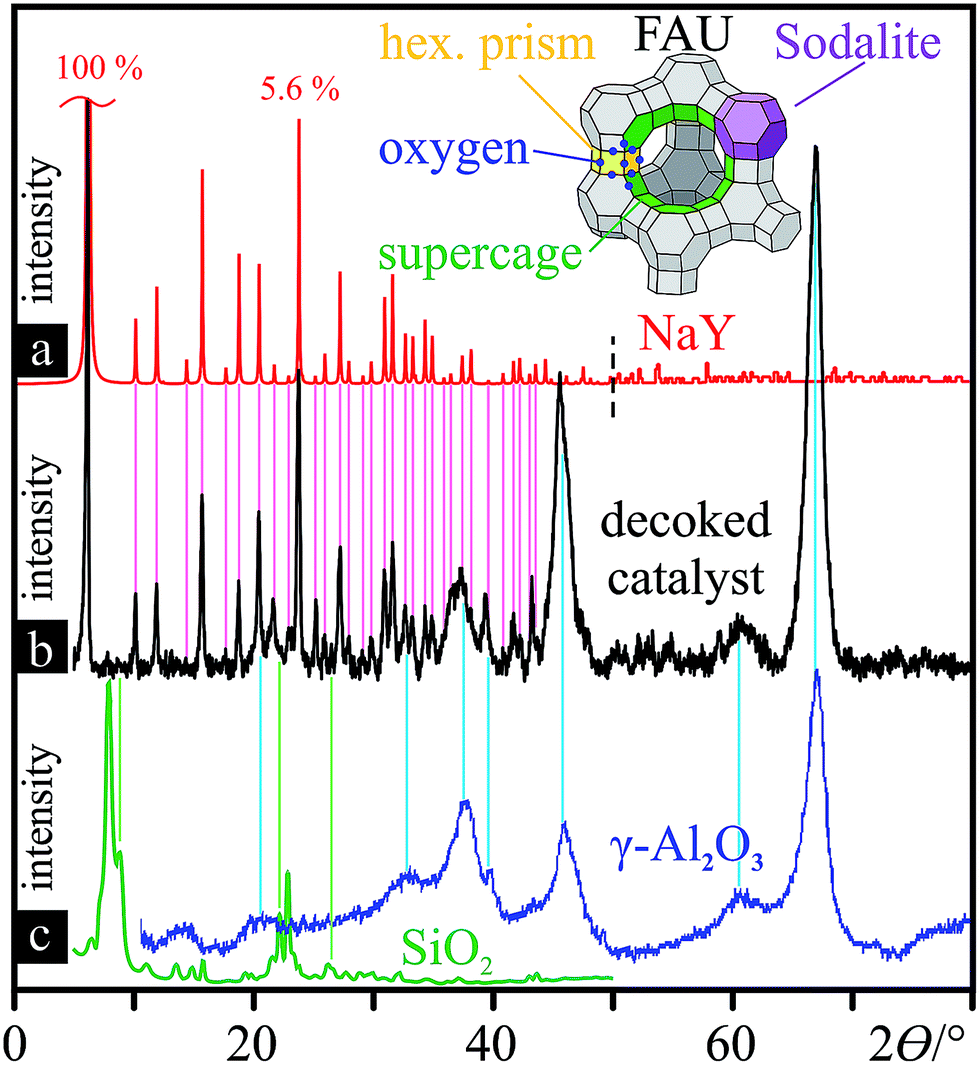 | ||
| Fig. 3 Decoked catalyst powder characterisation: XRD patterns with reference lines for peak assignment. (a) simulated XRD powder pattern of NaY FAU from 0–50° (ref. 3) and from 50–80° (ref. 97) (both sections normalised to the strongest reflection at 6.19°); 3-D structure of simplified FAU unit cell (Si/Al vertices), composed of linked sodalite cells. (b) XRD powder pattern of the decoked catalyst. (c) XRD pattern of zeolite (ITQ-39)-derived SiO2 (left, green)99 and commercial γ-Al2O3 (blue, right)98 (both baseline-corrected). | ||
The phase chemistry of the coke deposit was provided by Raman spectroscopy (Fig. 4). The signal of the decoked catalyst powder was subtracted from that of the FC3R to reveal an approximate spectrum of the coke (not free of catalyst contributions with parasite peaks). The presumed graphitic nature of the coke shell was confirmed with characteristic D and G modes, and their overtones G′ and 2G.101 The thin peak width of G indicates relatively ordered graphite with few defects,102 confirmed by a defect density ID/IG of 0.65.103 However, the relatively intense and wide D mode attests to the disordered carbon and numerous aromatic species present,104 as can be expected from a by-product of petroleum refining. This is supported by a low G′ intensity and very low IG′/ID ratio (0.15), indicating low purity.103
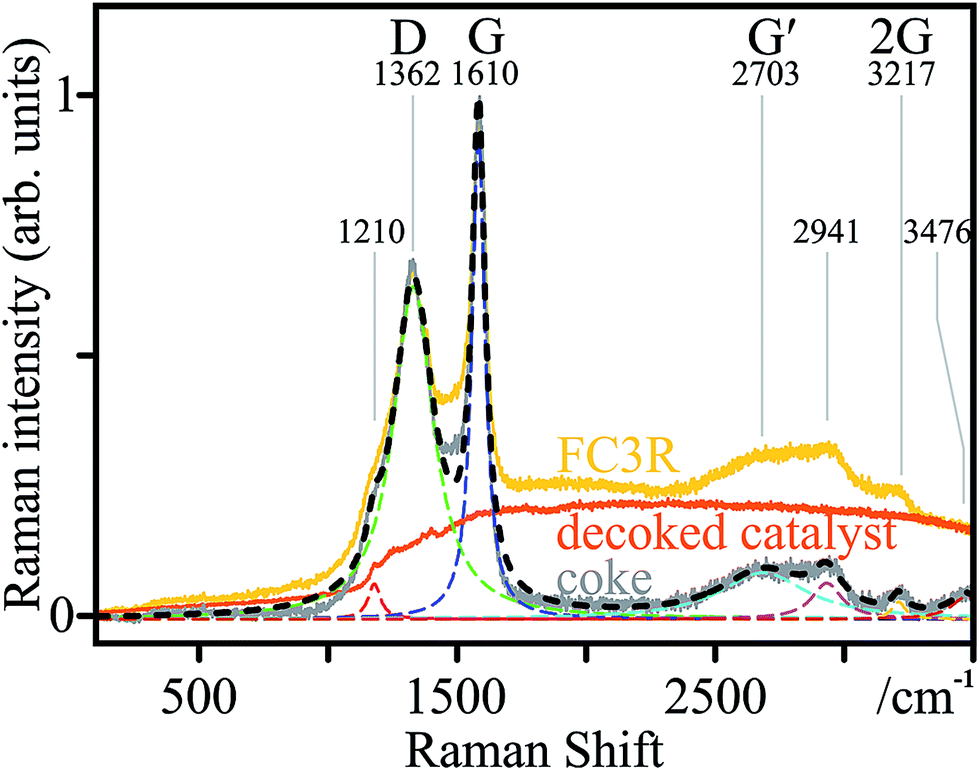 | ||
| Fig. 4 Coke characterisation: Raman spectrum (black) of the coke constructed (r2 > 0.98) from the deconvoluted peaks of the coke spectrum (grey), in turn resulting from FC3R (raw data; yellow) minus the decoked catalyst baseline (orange); marked peaks D, G, and their overtones G′ and 2G, and corresponding frequencies. Fits are dashed. | ||
3.2 SiONWs
Post synthesis, the thin film sample from Z1 (Fig. 1) appeared to be lacking any deposition or Ni film. In the SEM, except for a few protruding, torus-shaped craters, the entire sample surface was covered in a dense, regular layer of nanowires (Fig. 5a), found to be optically transparent. Judging from their shape, the craters might be due to larger target debris, projected at the beginning of the laser irradiation or impurities on the Ni surface, already present before irradiation. Upon closer inspection (of either feature) (Fig. 5b), arrangements consisting of radially-grown nanowires appear, which, when observed with primary electrons, reveal particles at their core (Fig. 5c). Higher magnification (Fig. 5d and e) confirms this radial configuration of densely packed nanowires around particles, of which a local EDS spectrum reveals a composition of predominant Ni, along with O, Si and P (Fig. 5f): a first indication of Ni-particle-bound SiONWs. These were identical to those in and around the craters. | ||
| Fig. 5 SiONW characterisation I: (a–e) SEM micrographs (high-energy secondary electrons in (c)) with increasing magnification; (f) EDS spectrum of the growth nucleus (point 1 in e). | ||
The bright heterostructures seen by SEM appear as transparent nanowires protruding from opaque particles using TEM (Fig. 6a). High magnification of the wires (Fig. 6b) exposed an amorphous structure confirmed by the absence of diffraction patterns (inset). A zoom on one nanowire/core formation (Fig. 6c) was used for elemental analysis: Ni is only found at the core (Fig. 6d), where Si (Fig. 6e) and O (Fig. 6f) are also present, yet to a greater extent at the surface and in the wires. The excess O, deduced from a corresponding Si/O atomic ratio of 1:3.5, possibly indicates Ni oxide in the core. Also identified were low amounts of P, in the core, and generally dispersed traces of Al, not corresponding to any particular structural feature or location.
 | ||
| Fig. 6 SiONW characterisation II: TEM micrographs of (a) Ni particles with radial SiONWs; (b) high magnification of a SiONW with diffraction inset; (c) close up from “a” of one Ni particle with SiONWs; (d–f) EDS scans of “c” for Ni, Si and O with 1:3.5 Si to O atomic ratio. | ||
These arrangements with catalyst cores suggest dewetting of the Ni thin film, a common occurrence in supported nanowire/-tube synthesis, by which a continuous thin film at or near its melting point ruptures and coagulates into beads, due to Plateau–Rayleigh instability and surface tension. Although 1200 °C is still well below the melting point of Ni (1450 °C), several factors can account for this phenomenon: (1) low dimensionality is known to drastically reduce melting points, placing that of a 100 nm Ni film at roughly 1200 °C;105 (2) the highly energetic species landing on the Ni, produced in the ablation plume (above 3500 °C (ref. 106)), would further provide the necessary energy; (3) the eutectic temperature of a formed alloy, e.g. the Ni–Si system, which has a number of eutectic plateaus between 22 and 56 at% Si, at temperatures as low as 800 °C (up to 1215 °C),107 all of which are allowed systems through Si diffusion. Any combination of (1), (2) and (3) would drastically reduce the melting temperature of the catalyst and initiate or maintain liquid metal-alloy droplets catalysing nanowire growth.
To explore dewetting, layer morphology, growth and possibly the early stages thereof, a deeper inspection of the SiONW layer down to the substrate was necessary. Since this layer was densely packed, a FIB cross-section provided the insight (Fig. 7). Although the Pt layer (see Experimental) presumably compressed the SiONW layer, a ∼ 6–7 μm layer of SiONWs (around Ni cores) can be seen atop an intact SiO thin film on Si, with no trace of the Ni layer. The nanowires seem to be thinner near the substrate. Furthermore, although Si- and SiONWs are often reported to coexist, growing with or from each other,86,87,108,109 no SiNWs were observed. Several conclusions can be drawn: (1) the existing SiO film did not intervene in the nanowire growth; (2) only SiONWs were grown; (3) the Ni layer was completely consumed into beads, and (4) nanowire growth in the lowest regions was quenched due to lacking precursors.
 | ||
| Fig. 7 SiONW layer: SEM micrography of a FIB cross-section, along the sample surface normal. Observed are (from top to bottom): an irregular layer of deposited platinum (partially sunken in), a ∼ 6–7 μm layer of SiONWs around Ni cores, an intact 250 nm SiO thin film (as received), and the underlying Si. The right of the image is in false colour, to better identify each species: pink Pt, green nanowires, blue cores and yellow SiO2 thin film. The vertical lines are an artefact of the FIB milling. | ||
The vapour–liquid–solid (VLS) mechanism would imply an initial downwards growth of the nanowires, elevating the first seeds of growth away from the substrate, eventually covering the space below. The silica, being deposited on the outer surface, would feed the continued growth of thick and dense nanowire bundles in the uppermost regions. However, the resulting limited availability of silica and Ni in the lower regions of the layer would result in smaller wires nucleating from small amounts of leftover metal catalyst, which was finally exhausted in the early stages of growth.
This complex SiONW precursor, FC3R, offers many conjectures as to the growth mechanism. Aside from the common vapour/solid–liquid–solid (SLS) mechanisms involving Si and O, certain reports present carbon as a key element in their SiONW growth, through mechanisms fitting the conditions described here. Upstream graphite can be gasified by a fed oxygen flow to CO2, reacting with Si (s) to produce SiO (g), which upon reaction with O2 (g) to SiO2 (s) in Si, aggregates into nanowires.110 Similarly, a CH4 feed can be decomposed by an Fe catalyst, freeing C to dissolve in SiO2 nanoparticles, forming SiO and CO vapours; CO reduction by the Fe releases O2 which diffuses with SiO in the nanoparticles to nucleate at their surface as SiONWs.111 In either case, the carbon was key and acted as a vehicle for the oxygen, to oxidise Si and SiO.
Summarising, the liquid catalyst droplets become the support for the described VLS growth, prior to or upon laser irradiation of the target. At this point, the reactants from the vapour phase are absorbed, alloyed and solved until supersaturation and crystallisation at the surface. Each nickel particle catalyses tens to thousands of nanowires depending on its size, maintained as long as the eutectic temperature is not underrun. Supposing the temperature of the catalyst is well above the chamber temperature of 1200 °C, due to the incoming vapour, all eutectic Ni–Si systems would be allowed.
As to the alumina present in the target, it appears to not play a role in the synthesis. Indeed, FAU thermally decomposes at 793 °C,93 freeing up mainly silica and alumina. Their respective melting and boiling temperatures of 1713 °C & 2950 °C (ref. 112) vs. 2072 °C & 2977 °C (ref. 113) would indicate a tendency to preferentially release the silica into the laser plume. Furthermore, the heavier alumina (4.0–4.1 vs. 2.2–2.6 g cm−3) would have a shorter flight path from the target and perhaps land outside of the collector. Finally, FAUs X and Y and other variations differ mainly in the Si/Al ratio. It is possible that this was a low-Al FAU and that the kaolinite γ-Al2O3 underwent different ejection/vaporisation mechanisms, incompatible with nanostructure self-assembly. Si, found in all the catalyst components (FAU, binder, kaolinite), was in major surplus with respect to Al (kaolinite and minor component of FAU). Although bottom-up synthesis of alumina nanowires is known to occur at the reactor temperature (1200 °C) by PVD114 and CVD,115 no such reports exist for PLD and none were observed here.
3.3 SWCNTs
Neither SEM, TEM nor EDS analyses indicated CNTs in samples from zone Z1. SEM EDS spectra of what appeared to be thinner nanowires/-tubes indicated low amounts of carbon, yet within uncertain peak intensities. However, Raman spectra from certain spots of the sample (Fig. 8-Z1) revealed spectra strikingly similar to that of MWCNTs, with much thinner peaks, a high D-mode intensity, a slightly lower ID/IG of 0.61 and, most importantly, a very intense G′ and thus a much lower IG′/ID of 0.92, common in MWCNTs. Additionally, the refined Raman modes of said spectrum attest to the loss of amorphous carbon, aromatic species and functional groups, previously present in the coke.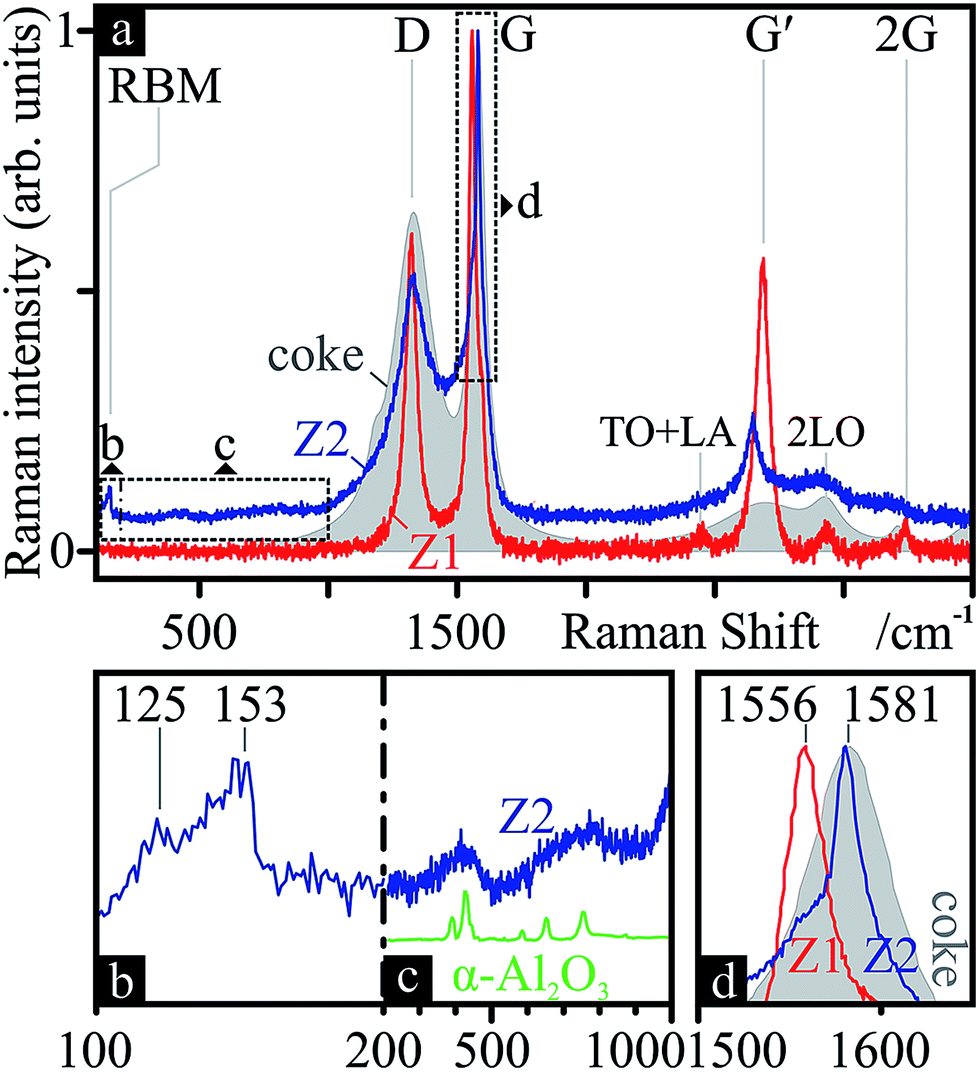 | ||
| Fig. 8 Nanocarbon chemistry: Raman spectra (a) of certain areas on the SiONW sample from zone Z1 (Z1, red) and of the black soot powder from zone Z2 (Z2, blue). The underlayed Raman spectrum of the initial coke from Fig. 4 (grey, filled area) is added for comparison. The RBM (b), alumina (c) (contrasted with ref. 124 in green) and G (d) regions are zoomed-in for further detail. Modes and frequencies are marked. | ||
In zone Z2 of the PLD apparatus, a black, powdery soot condensed on the cold tube exit. These samples produced Raman spectra (Fig. 8-Z2) indicative of graphitic species, including SWCNTs: a clear RBM (Fig. 8b) between 125 and 153 cm−1, revealing tubes ca. 1.5–1.8 nm in diameter;116 and G-splitting (Fig. 8d), with disparate G+ and G− intensities, suggesting semiconducting SWCNTs.117 The position of the G band (1581 cm−1) suggests a more ordered form of sp2 carbon than that found in Z1 (1561 cm−1).118 Furthermore, the Raman features between RBM and D (Fig. 8c) resemble that of α-Al2O3. This γ → (δ → θ →) α transformation is to be expected, since the former is a metastable phase and only α remains at 1100 °C.119
EDS spectra (Fig. 9b) reveal the same components as the nanowire samples, C, O, Al and Si, in order of atomic abundance. Although C is predominant, the missing Al in zone Z1 clearly made its way here, accounting for a quarter of the atomic species. Indeed, the bright spots in Fig. 9a appear to be alumina particles (Fig. 9c). Apart from these visible spheres, Si, Al and O are present throughout the sample (Fig. 9b). Closer up, branched networks of nanoparticulate material appear (Fig. 9d), typical for CNT soot.
 | ||
| Fig. 9 Nanocarbon soot morphology and chemistry: SEM (a) of powder deposit from zone Z2 in the PLD apparatus (Fig. 1) with corresponding EDS scan (b); SEM with inset EDS scans of an alumina particle (c); higher magnification of branched network (d). | ||
The latter formations, upon observation in TEM (Fig. 10), reveal superimposed species. SiC nanoparticles (Fig. 10b) and -flakes (Fig. 10c), encapsulated with 2–3 layers of sp2 carbon display a characteristic honeycomb pattern with a fringe lattice spacing of 0.28 nm (confirmed by the inset fast Fourier transform (FFT)).120 OLCs (Fig. 10d) and CNDs (Fig. 10e) are also present. Most importantly, SWCNTs (Fig. 10e) are readily found, as an underlying network and protruding from the edges of CNDs/SiC, with diameters >2 nm, undetectable by Raman.121 Conversely, those observed by Raman, 1.5–1.8 nm (125–153 cm−1),116 could not be found by TEM, likely hidden amongst the other species present. Although MWCNTs with inner-most tubes below 2 nm are known to present misleading RBM121,122 and G-splitting121 Raman features, none could be observed.
 | ||
| Fig. 10 Nanocarbon soot (a) species: TEM micrographs with corresponding fringe lattice spacings of sp2-carbon-encapsulated SiC nanocrystals (b), a nanoflake (insets: zoomed-in edge and lattice; FFT) (c), an OLC (d) and SWCNTs (e) with encapsulated SiC or CNDs at their base. | ||
This CNT growth follows the mechanism from whence it was discovered: SLS-condensation on the downstream, cooled furnace exit.27 Though instead of a water-cooled copper collector, the air-cooled quartz tube served as condensation site. Specifically, gaseous carbon was dissolved in liquid SiC and C nanoparticles to saturation, followed by crystallisation at the surface. This precipitation of graphitic carbon at the surface encapsulates the nanoparticle in sp2 C. Through an instability in the sheet or partial dissolution of fullerenes, critical nuclei protrude outwards into CNTs.38 Failure to produce outwards growth, closes the carbon shell resulting in the observed encapsulated SiC, CNDs or OLCs.
Although the 47% void fraction of FAU123 is actually conducive to enhanced CNT synthesis and higher yields by PLD due to porosity,26 these pores likely contain oxygen. This presents competing mechanisms of amorphous carbon removal towards increased CNT purity and the removal of carbonaceous species before it can be added to the CNT. Still, contrary to graphite ablation, where single atoms are added to a growing CNT, larger aromatic species present in the coke can be directly incorporated.39 Finally, no alumina nanoparticulate could be found, crystalline or other.
The benefits of the low cost and availability of FC3R outweigh the complex heterogeneous composition of the coke, with further research necessary to fine-tune the selectivity, quality and yield of the numerous valuable products. The advantages of PLD with respect to its plume dynamics could be combined with the scalability of CVD, tailoring CNT seeds by the former with continued growth by the latter. Alternating functionally-graded CNT layers could be achieved by changing the pulse parameters.
The worlds refined hydrocarbons are chiefly produced by FCC and HP (48% and 19%).1 Extending these findings to spent cracking catalyst waste in general, the little-to-no silica precludes the use of spent HP catalyst for SiONWs; and although the metal content might be pertinent to SWCNT catalysis, the low amounts of carbon (due to a reduced coking tendency) and the absence of any tendency for alumina to self-assemble in an oven-laser apparatus, would render HPCR a mere source of alumina nanoparticulate with marginal SWCNT yield, at best. Thus, FC3R stands out as a candidate for revalorisation as proposed here.
4. Conclusions
The waste, taken directly from a refinery, was identified as high-carbon petrochemical cracking catalyst waste, FC3R, consisting of petroleum coke-covered (predominantly sp2) zeolite Y and γ-Al2O3. The resulting PLD nanospecies were produced in two distinct zones of a simple lab-scale oven-laser setup, from untreated FC3R. On samples directly opposite the FC3R target (zone Z1), the reflected plume produced an optically transparent layer of amorphous SiONWs, consistent with a metal-catalysed VLS mechanism, as densely packed, radial bundles around Ni particle cores at their base. At the downstream, cooled tube-furnace exit (zone Z2), condensation produced SWCNTs (and other nanocarbons: sp2-encapsulated SiC nanocrystals and -flakes, OLCs and CNDs), consistent with a nanocarbon-catalysed SLS self-assembly. Our hypothesis and concept are thus proven, constituting (1) the first use of a waste feedstock for SiONW synthesis, (2) one of very few such attempts for CNTs, and (3) the first (high-tech) outlet for FC3R and catalyst coke in general.Silicon is the most eminent material in electronics. From bulk to thin films, its semiconducting properties have permeated technology, until recently, unchallenged. Enter wires, tubes and dots of various materials. Thanks to their size and/or aspect ratio, these low-dimensional nanostructures are now competing for their share in future technologies. Carbon, from the same group in the periodic table, is possibly the strongest contender. Respectively the 2nd and 15th most abundant elements in earth's crust, neither Si nor C are scarce nor are they conflict materials. But as high-tech nanomaterials continue to infiltrate technology, so does the need to reduce, reuse and recycle (‘the three R's’). Although a petrochemical precursor may not qualify as green, its use does address the third R of a currently otherwise wasted and contaminant resource, mitigating its environmental impact. Moreover, if recycled materials are of particular relevance since they avoid waste and its disposal, then indeed transforming petroleum cracking waste into 0- and 1-D nanostructures of great technological value such as SWCNTs and SiONWs would fall into the more coveted end of the recycling spectrum, upcycling. This extreme revalorisation of abundant waste could fuel research and applications in mesoscopic physics, optical fibres, Si- and C-based nano- and microelectronic devices and composites.
Acknowledgements
This work was supported by the SUMA2 Network Project, 7th Framework Program of the European Commission (IRSES Project No. 318903), and the CREATE-Network Project, Horizon 2020 Program of the European Commission (RISE Project No. 644013). VP and MZ thank Prof. Eduard Arzt (INM) for his continuing support. AB was supported as part of the iSTAR Scholars Program of Drexel University.Notes and references
- E. T. C. Vogt and B. M. Weckhuysen, Chem. Soc. Rev., 2015, 44, 7342–7370 RSC.
- R. Sadeghbeige, Fluid Catalytic Cracking Handbook: Design, Operation and Troubleshooting of FCC Facilities, Gulf Professional Publishing, 2nd edn, 2000 Search PubMed.
- M. M. J. Treacy and J. B. Higgins, Collection of simulated XRD powder patterns for zeolites, Elsevier, 1986, vol. 21 Search PubMed.
- D. L. Trimm, Appl. Catal., A, 2001, 212, 153–160 CrossRef CAS.
- A. L. C. da Cunha, M. S. Lemos, S. Meth, J. P. Gonçalves and J. Dweck, J. Therm. Anal. Calorim., 2011, 106, 805–809 CrossRef.
- N. Su, H.-Y. Fang, Z.-H. Chen and F.-S. Liu, Cem. Concr. Res., 2000, 30, 1773–1783 CrossRef CAS.
- E. Furimsky, Catal. Today, 1996, 30, 223–286 CrossRef CAS.
- U.S. Energy Information Administration, Supply and Disposition – Petroleum Coke, 2015 Search PubMed.
- M. A. Díez, R. Alvarez and C. Barriocanal, Int. J. Coal Geol., 2002, 50, 389–412 CrossRef.
- R. A. Graves Jr and W. G. Witte, Flight-test analysis of Apollo heat-shield material using the Pacemaker vehicle system, Langley Research Center, Washington DC, 1968 Search PubMed.
- European Commision, Integr. Pollut. Prev. Control, 2003, p. 518 Search PubMed.
- E. C. I. Council, FCC equilibrium catalyst (including FCC catalyst fines) finds safe reuse/rework outlets in Europe, 2006 Search PubMed.
- M. Diaz, Hazardous Waste Management System; Identification and Listing of Hazardous Waste: Spent Catalysts from Dual-Purpose Petroleum Hydroprocessing Reactors, Washington DC, 2002, vol. 67 Search PubMed.
- Environmental and forestry Statistics, Guidance on classification of waste according to EWC-stat categories, 2010, vol. 2150/2002 Search PubMed.
- M. Marafi and A. Stanislaus, Resour., Conserv. Recycl., 2008, 53, 1–26 CrossRef.
- R. Schmitt, Oil Gas J., 1991, 89, 101–102 CAS.
- J. Monzó Balbuena, J. Payá Bernabeu and V. Rosado Borrachero, Sp. Pat., ES 2 144 928, 1997.
- E. Restrepo, L. Arbeláez, P. Caballero, E. López, F. Vargas and G. Duarte, Elabor. refract. Al2O3–SiO2 a partir residuos catalíticos provenientes la Ind. petroquímica, 2014, pp. 1–10 Search PubMed.
- L. P. Garcia, R. T. da Cruz and S. R. Braganca, Proc. Inst. Mech. Eng., Part L, 2012, 226, 286–292 CAS.
- A. Escardino, J. L. Amoros, A. Moreno and E. Sanchez, Waste Manage. Res., 1995, 13, 569–578 CrossRef CAS.
- K.-L. Lin, H.-H. Wu, S.-J. Chao, A. Cheng and C.-L. Hwang, Environ. Prog. Sustainable Energy, 2013, 32, 94–98 CrossRef CAS.
- J. Monzó, J. Payá, M. V. Borrachero, S. Velásquez, L. Soriano, P. Serna and J. Rigueira, in International RILEM Conference on the Use of Recycled Materials in Buildings and Structures, 2004 Search PubMed.
- A. Guerrero, J. Puerta, F. Gomez and F. Blanco, Phys. Scr., 2008, T131, 014007 CrossRef.
- N. Nagaraju, A. Fonseca, Z. Konya and J. B. Nagy, J. Mol. Catal. A: Chem., 2002, 181, 57–62 CrossRef CAS.
- K.-H. Lee, K. Baik, J.-S. Bang, S.-W. Lee and W. Sigmund, Solid State Commun., 2004, 129, 583–587 CrossRef CAS.
- M. Yudasaka, M. Zhang and S. Iijima, Chem. Phys. Lett., 2000, 6–7 Search PubMed.
- T. Guo, P. Nikolaev and A. Thess, Chem. Phys. Lett., 1995, 243, 49–54 CrossRef CAS.
- H. Zhu, L. Ci, C. Xu, J. Liang and D. Wu, Diamond Relat. Mater., 2002, 11, 1349–1352 CrossRef CAS.
- D. P. Yu, Q. L. Hang, Y. Ding, H. Z. Zhang, Z. G. Bai, J. J. Wang, Y. H. Zou, W. Qian, G. C. Xiong and S. Q. Feng, Appl. Phys. Lett., 1998, 73, 3076–3078 CrossRef CAS.
- J. Chen, X. Li, X. Wu, J. T. Pierce, N. Fahruddin, M. Wu and J. X. Zhao, Langmuir, 2014, 30, 9514–9523 CrossRef CAS PubMed.
- E. Huey, S. Krishnan, S. K. Arya, A. Dey and S. Bhansali, Sens. Actuators, B, 2012, 175, 29–33 CrossRef CAS.
- S. Y. Khmel, in EU PVSEC, 2013, pp. 395–398 Search PubMed.
- N. S. Ramgir, P. K. Sekhar, A. Zajac, L. Lee, T. Zhukov and S. Bhansali, Sens. Lett., 2007, 5, 608–611 CrossRef CAS.
- R. G. Elliman, A. R. Wilkinson, T. H. Kim, P. K. Sekhar and S. Bhansali, J. Appl. Phys., 2008, 103, 104304 CrossRef.
- G. Brambilla and D. N. Payne, Nano Lett., 2009, 9, 831–835 CrossRef CAS PubMed.
- T. Guo, P. Nikolaev, A. G. Rinzler, D. Tomanek, D. T. Colbert and R. E. Smalley, J. Phys. Chem., 1995, 99, 10694–10697 CrossRef CAS.
- E. Munoz, W. Maser and A. Benito, Synth. Met., 1999, 103, 2490–2491 CrossRef CAS.
- A. Gorbunoff and O. Jost, in Pulsed Laser Deposition of Thin Films, 2007, pp. 613–632 Search PubMed.
- L. Pang, L. Prochazka and R. Quezada, Fuel and Energy Abstracts, 1995, 36, 189 Search PubMed.
- G. Hong, Y. Chen, P. Li and J. Zhang, Carbon, 2012, 50, 2067–2082 CrossRef CAS.
- D. Carole, A. Brioude, A. Pillonnet, J. Lorenzzi, O. Kim-Hak, F. Cauwet and G. Ferro, J. Cryst. Growth, 2011, 320, 55–62 CrossRef CAS.
- M. Sharon, W. K. Hsu, H. W. Kroto, D. R. M. Walton, A. Kawahara, T. Ishihara and Y. Takita, J. Power Sources, 2002, 104, 148–153 CrossRef CAS.
- M. Kumar and Y. Ando, Diamond Relat. Mater., 2003, 12, 998–1002 CrossRef CAS.
- M. Kumar and Y. Ando, Diamond Relat. Mater., 2003, 12, 1845–1850 CrossRef CAS.
- M. Kumar and Y. Ando, Chem. Phys. Lett., 2003, 374, 521–526 CrossRef CAS.
- M. Kumar and Y. Ando, Carbon, 2005, 43, 533–540 CrossRef CAS.
- R. J. Andrews, C. F. Smith and A. J. Alexander, Carbon, 2006, 44, 341–347 CrossRef CAS.
- M. Kumar, T. Okazaki, M. Hiramatsu and Y. Ando, Carbon, 2007, 45, 1899–1904 CrossRef CAS.
- R. A. Afre, T. Soga, T. Jimbo, M. Kumar, Y. Ando, M. Sharon, P. R. Somani and M. Umeno, Microporous Mesoporous Mater., 2006, 96, 184–190 CrossRef CAS.
- P. Ghosh, T. Soga, R. A. Afre and T. Jimbo, J. Alloys Compd., 2008, 462, 289–293 CrossRef CAS.
- P. Ghosh, R. A. Afre, T. Soga and T. Jimbo, Mater. Lett., 2007, 61, 3768–3770 CrossRef CAS.
- A. B. Suriani, R. M. Nor and M. Rusop, J. Ceram. Soc. Jpn., 2010, 118, 963–968 CrossRef CAS.
- L. Pang and M. Wilson, Energy Fuels, 1993, 239–240 CAS.
- K. Williams, M. Tachibana and J. Allen, Chem. Phys., 1999, 31–37 CAS.
- M. Wilson, H. Patney and J. Kalman, Fuel, 2002, 81, 5–14 CrossRef CAS.
- J.-S. Qiu, F. Zhang, Y. Zhou, H.-M. Han, D.-S. Hu, S. C. Tsang and P. J. F. Harris, Fuel, 2002, 81, 1509–1514 CrossRef CAS.
- Y. F. Li, J. S. Qiu, Z. B. Zhao, T. H. Wang, Y. P. Wang and W. Li, Chem. Phys. Lett., 2002, 366, 544–550 CrossRef CAS.
- J. Qiu, Y. Li, Y. Wang, T. Wang, Z. Zhao and Y. Zhou, Carbon, 2003, 41, 2170–2173 CrossRef CAS.
- J. Yu, J. Lucas, V. Strezov and T. Wall, Fuel, 2003, 82, 2025–2032 CrossRef CAS.
- Y. J. Tian, Y. L. Zhang, Q. Yü, X. Z. Wang, Z. Hu, Y. F. Zhang and K. C. Xie, Catal. Today, 2004, 89, 233–236 CrossRef CAS.
- J. Qiu, Y. An, Z. Zhao, Y. Li and Y. Zhou, Fuel Process. Technol., 2004, 85, 913–920 CrossRef CAS.
- J. Qiu, Y. Li, Y. Wang and W. Li, Fuel Process. Technol., 2004, 85, 1663–1670 CrossRef CAS.
- Y. Tian, Y. Zhang, B. Wang, W. Ji, Y. Zhang and K. Xie, Carbon, 2004, 42, 2597–2601 CrossRef CAS.
- Z. Wang, Z. Zhao and J. Qiu, Carbon, 2006, 44, 1321–1324 CrossRef CAS.
- Z. Wang, Z. Zhao and J. Qiu, Carbon, 2006, 44, 1845–1847 CrossRef CAS.
- J. Qiu, Q. Li, Z. Wang, Y. Sun and H. Zhang, Carbon, 2006, 44, 2565–2568 CrossRef CAS.
- R. B. Mathur, C. Lal and D. K. Sharma, Energy Sources, Part A, 2007, 29, 21–27 CrossRef CAS.
- J. Qiu, Z. Wang, Z. Zhao and T. Wang, Fuel, 2007, 86, 282–286 CrossRef CAS.
- Y. Zhang and S. Iijima, Appl. Phys. Lett., 1999, 75, 3087 CrossRef CAS.
- V. Lavrentiev, H. Abe and S. Yamamoto, Phys. B, 2002, 323, 303–305 CrossRef CAS.
- J. Qiu, G. Chen, Z. Li and Z. Zhao, Carbon, 2010, 48, 1312–1315 CrossRef CAS.
- M. Zhang, M. Yudasaka and S. Iijima, Chem. Phys. Lett., 2001, 336, 196–200 CrossRef CAS.
- D. B. Geohegan, H. Schittenhelm, X. Fan, S. J. Pennycook, A. A. Puretzky, M. A. Guillorn, D. A. Blom and D. C. Joy, Appl. Phys. Lett., 2001, 78, 3307 CrossRef CAS.
- A. A. Stramel, M. C. Gupta, H. R. Lee, J. Yu and W. C. Edwards, Optic. Laser. Eng., 2010, 48, 1291–1295 CrossRef.
- L. Li, F. Li, C. Liu and H.-M. Cheng, Carbon, 2005, 43, 623–629 CrossRef CAS.
- F. Bonaccorso, C. Bongiorno, B. Fazio, P. G. Gucciardi, O. M. Maragò, A. Morone and C. Spinella, Appl. Surf. Sci., 2007, 254, 1260–1263 CrossRef CAS.
- Y. Li, H. Wang, G. Wang and J. Gao, Chem. Eng. J., 2012, 211–212, 255–259 CrossRef CAS.
- L. E. Murr, D. K. Brown, E. V. Esquivel, T. D. Ponda, F. Martinez and A. Virgen, Mater. Charact., 2005, 55, 371–377 CrossRef CAS.
- H. M. Yusoff, R. Shastry, T. Querrioux and J. Abrahamson, Curr. Appl. Phys., 2006, 6, 422–426 CrossRef.
- D. Buchholz, S. Doherty and R. Chang, Carbon, 2003, 41, 1625–1634 CrossRef CAS.
- K. S. Kim, G. Cota-Sanchez, C. T. Kingston, M. Imris, B. Simard and G. Soucy, J. Phys. D: Appl. Phys., 2007, 40, 2375–2387 CrossRef CAS.
- K. Xu, Y. Li, C. Xu, J. Gao, H. Liu, H. Yang and P. Richard, Chem. Eng. J., 2013, 225, 210–215 CrossRef CAS.
- K. Xu, Y. Li, T. Liu, F. Yang, C. Xu and J. Gao, IEEE Nanotechnology Magazine, 2013, 15–18 CrossRef.
- K. Xu, Y. Li, F. Yang, W. Yang, L. Zhang, C. Xu, T. Kaneko and R. Hatakeyama, Carbon, 2014, 68, 511–519 CrossRef CAS.
- S. Abdullayeva and N. Musayeva, World J. Condens. Matter Phys., 2014, 93–100 CrossRef.
- C. P. Li, X. H. Sun, N. B. Wong, C. S. Lee, S. T. Lee and B. K. Teo, Chem. Phys. Lett., 2002, 365, 22–26 CrossRef CAS.
- B. K. Teo, C. P. Li, X. H. Sun, N. B. Wong and S. T. Lee, Inorg. Chem., 2003, 42, 6723–6728 CrossRef CAS PubMed.
- A. Bazargan and G. McKay, Chem. Eng. J., 2012, 195–196, 377–391 CrossRef CAS.
- W. Zhao, B. Basnet and I. J. Kim, J. Adv. Ceram., 2012, 1, 179–193 CrossRef CAS.
- N. Wang, Nature, 2000, 408, 50–51 CrossRef CAS PubMed.
- Plan Director de Residuos Sólidos de Montevideo y Área Metropolitana - ANEXO: Residuos Sólidos Industriales, http://www.cempre.org.uy/docs/biblioteca/plan_director/etapa1_estudios_basicos/Anexo_RSI.PDF, (accessed June 2016), Fichtner GmbH CO&KG and LKSur S.A., 2005 Search PubMed.
- N. Souza, M. Zeiger, V. Presser and F. Mücklich, RSC Adv., 2015, 5, 62149–62159 RSC.
- S. Bhatia, Zeolite catalysis: principles and applications, CRC Press, Boca Raton, 1990 Search PubMed.
- W. A. Deer and R. A. Howie, Rock-forming Minerals: Silica Minerals, Geological Society of London, London, 2004 Search PubMed.
- W. N. Reynolds, Physical properties of graphite, Elsevier, 1968 Search PubMed.
- C. S. Cundy and P. A. Cox, Microporous Mesoporous Mater., 2005, 82, 1–78 CrossRef CAS.
- C. Baerlocher and L. B. McCusker, Database zeolite struct., http://www.iza-structure.org Search PubMed.
- N. Salahudeen, A. S. Ahmed, A. H. Al-Muhtaseb, M. Dauda, S. M. Waziri and B. Y. Jibril, Appl. Clay Sci., 2015, 105–106, 170–177 CrossRef CAS.
- T. Willhammar, J. Sun, W. Wan, P. Oleynikov, D. Zhang, X. Zou, M. Moliner, J. Gonzalez, C. Martínez, F. Rey and A. Corma, Nat. Chem., 2012, 4, 188–194 CrossRef CAS PubMed.
- A. L. Patterson, Phys. Rev., 1939, 56, 978–982 CrossRef CAS.
- S. Reich and C. Thomsen, Philos. Trans. R. Soc., A, 2004, 362, 2271–2288 CrossRef CAS PubMed.
- L. Ian, Handbook of Raman Spectroscopy. From the Research Laboratory to the Process Line, CRC Press, New York and Basel, 2001, vol. 28 Search PubMed.
- R. A. DiLeo, B. J. Landi and R. P. Raffaelle, J. Appl. Phys., 2007, 101, 064307 CrossRef.
- J. Schwan, S. Ulrich, V. Batori, H. Ehrhardt and S. R. P. Silva, J. Appl. Phys., 1996, 80, 440 CrossRef CAS.
- D. G. Gromov and S. A. Gavrilov, Thermodynamics – Physical Chemistry of Aqueous Systems, InTech, 2011 Search PubMed.
- K. Pathak, PhD thesis, University of Akron, 2008.
- P. Nash and A. Nash, Bull. Alloy Phase Diagrams, 1987, 8, 6–14 CrossRef CAS.
- Y. Chen, J. Li and J. Dai, Chem. Phys. Lett., 2001, 344, 450–456 CrossRef CAS.
- H. Wang, X. Zhang, X. Meng, S. Zhou, S. Wu, W. Shi and S. Lee, Angew. Chem., Int. Ed., 2005, 44, 6934–6937 CrossRef CAS PubMed.
- K. Saulig-Wenger, D. Cornu, F. Chassagneux, T. Epicier and P. Miele, J. Mater. Chem., 2003, 13, 3058 RSC.
- K. H. Lee, S. W. Lee, R. R. Vanfleet and W. Sigmund, Chem. Phys. Lett., 2003, 376, 498–503 CrossRef CAS.
- D. R. Lide, Handbook of Chemistry and Physics, CRC Press, 2002 Search PubMed.
- P. Patnaik, Handbook of Inorganic Chemicals, 2003 Search PubMed.
- Q. Zhao, X. Xu, H. Zhang, Y. Chen, J. Xu and D. Yu, Appl. Phys. A: Mater. Sci. Process., 2004, 79, 1721–1724 CAS.
- S. J. Park, J. G. Kim and D. J. Choi, Int. J. Appl. Ceram. Technol., 2015, 6, 1–6 Search PubMed.
- C. Fantini, A. Jorio, M. Souza, M. S. Strano, M. S. Dresselhaus and M. A. Pimenta, Phys. Rev. Lett., 2004, 93, 1–4 Search PubMed.
- M. S. Dresselhaus, G. Dresselhaus, R. Saito and A. Jorio, Phys. Rep., 2005, 409, 47–99 CrossRef.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- P. S. Santos, H. S. Santos and S. P. Toledo, Mater. Res., 2000, 3, 104–114 CAS.
- S. S. Lin, J. Phys. Chem. C, 2012, 116, 3951–3955 CAS.
- J. H. Lehman, M. Terrones, E. Mansfield, K. E. Hurst and V. Meunier, Carbon, 2011, 49, 2581–2602 CrossRef CAS.
- R. Gupta, B. P. Singh, V. N. Singh, T. K. Gupta and R. B. Mathur, Carbon, 2014, 66, 724–726 CrossRef CAS.
- D. W. Breck, Zeolite molecular sieves: structure, chemistry, and use, Wiley, 99th edn, 1973 Search PubMed.
- D. Zappa, S. Dalola, G. Faglia, E. Comini, M. Ferroni, C. Soldano, V. Ferrari and G. Sberveglieri, Beilstein J. Nanotechnol., 2014, 5, 927–936 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2016 |