
Shell thickness effects on reconfiguration of NiOcore–Ptshell anodic catalysts in a high current density direct methanol fuel cell†
Abstract
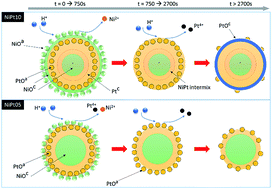
The atomic structure relocation of a NiOcore–Ptshell nanocatalyst (NC) in direct methanol fuel cells for a long-term work cycle is quantitatively investigated by performing both X-ray absorption spectroscopy and electrochemical potential sweeping analyses. We found that Pt atoms in this metal-to-metal oxide junction NC tend to dissolute on reducing the domain size in a long-term DMFC work cycle in the condition of Pt > 30 at%. On the other hand, with Pt < 30%, Pt atoms are rearranged into a thin homoatomic shell at the NiO surface. Such an atomic reconfiguration is controlled by competition between dissolution and relocation of Pt/Ni atoms at varying facets. For the optimum case (Pt < 30%), the current density and the output power density of DMFC are doubled by using NiO–Ptshell NC at the anode as compared to that using a PtRu alloy.
 Please wait while we load your content...
Please wait while we load your content...

