
Organic–inorganic hybrid polyoxometalate and its graphene oxide–Fe3O4 nanocomposite, synthesis, characterization and their applications as nanocatalysts for the Knoevenagel condensation and the synthesis of 2,3-dihydroquinazolin-4(1H)-ones†
Roushan Khoshnavazi*,
Leila Bahrami and
Forugh Havasi
University of Kurdistan, The Islamic Republic of Iran. E-mail: r.khoshnavazi@uok.ac.ir
First published on 14th October 2016
Abstract
The simple and high yield synthesis of H6Cu2[PPD]6[SiW9Cu3O37]·12H2O (PPD = p-phenylenediamine) as a new organic–inorganic hybrid polyoxometalate is reported. The new compound consists of trimetal-substitution polyoxometalate of A-β-SiW9Cu3O3710−, water molecules and coordinated and noncoordinated PPD molecules. H6Cu2[PPD]6[SiW9Cu3O37]·12H2O was firmly grafted on graphene oxide decorated with Fe3O4 nanoparticles. These nanocomposites were characterized by elemental analyses, thermogravimetric analysis (TGA), Fourier transform infrared (FT-IR), X-ray photoelectron spectrum (XPS), powder X-ray diffraction (XRD), scanning electron microscopy (SEM), energy dispersive X-ray analysis (EDX), and alternating gradient force magnetometer (AGFM). The results indicate that the size range of the nanoparticles are between 20 and 60 nm. The catalytic activity of H6Cu2[PPD]6[SiW9Cu3O37]·12H2O and the magnetically reusable nanocomposite of GO@Fe3O4@H6Cu2[PPD]6[SiW9Cu3O37]·12H2O were investigated in the Knoevenagel condensation and synthesis of 2,3-dihydroquinazolin-4(1H)-ones.
Introduction
The design of active, selective and recyclable heterogeneous catalysts based on polyoxometalates (POMs) have attracted great interest because of their controllable shape and size, highly negative charges and their numerous advantageous properties.1 Keggin-type POMs such as H3PMo12O40, H3PW12O40, and H4SiW12O40 (also known as heteropolyacids, HPAs) are strong Brønsted acid that make them useful as solid-acid catalysts for many acid catalyzed organic transformations.2–6 Despite the above advantages, application of POMs is restricted because of low surface area (<10 m2 g−1) and high solubility in water and polar organic solvents.7 Dispersion of POMs on a support such as silica,8 zeolite,9 or design of catalysts based on polyoxometalate nanoparticles10 can dissolve these problems and improve their catalytic activity.Functionalization of classical POMs by organic compounds with suitable functional groups not only provides a powerful way to gain multifarious new compounds but also affords a new method to modify catalytic performance of POMs.11 The design and synthesis of inorganic–organic hybrid materials based on POMs has received considerable attention due to their application in catalysis, sorption, and electrical conductivity.12,13 A wide variety of organic molecules have to be combined with POMs to explore the novel functional property and morphology of the type of inorganic–organic hybrid materials.14,15 Magnetic separation renders the recovery of catalysts from liquid-phase reactions much easier than by cross-flow filtration and centrifugation. It is simple, economical and promising for industrial applications.16 Among the various magnetic nanoparticles under investigation, Fe3O4 nanoparticles are arguably the most extensively studied as a core magnetic support because of their simple synthesis, low cost, and relatively large magnetic susceptibility of Fe3O4 SPNs.17 Thanks to its exceptionally high surface area (2630 m2 g−1) and abundant oxygenate groups such as epoxy, hydroxyl, and carboxyl groups18,19 on their surface, graphene oxide (GO) is as an ideal support for improving POMs catalytic activity.20 If the graphene oxide is loaded with Fe3O4 nanoparticles, the combined supporting material has key properties such as magnetic separation, the large surface area and ease of functionalizing with various chemical groups to increase their dependence toward target compounds.21 Heterogeneous catalysts such as graphene oxide–Fe3O4 have attracted a great deal of attention in recent years due to their high catalytic activity and interesting structures.22 The Fe3O4–GO magnetic nanoparticles have been most useful as heterogeneous catalysts because of their numerous applications in biotechnology, nanocatalysis, and medicine.23 In this paper, we report the synthesis and characterization of the new nanosized polyoxometalate-based inorganic–organic hybrid material, H6Cu2[PPD]6[SiW9Cu3O37]·12H2O (HybPOM) and the magnetically reusable nanocomposites of GO@Fe3O4@HybPOM as novel and efficient catalysts for synthesis of benzylidenemalononitrile and 2,3-dihydroquinazolin-4(1H)-one derivatives. The magnetic properties make the complete recovery of the catalyst possible by means of an external magnetic field.
Experimental
General
All reagents and solvents were commercially obtained from Merck chemical company and used without further purification. The Na9H[β-SiW9O34]·18H2O was prepared and characterized according to the method mentioned in the literature.24 Graphene oxide (GO) was prepared by modified Hummers' method.25 The C, H, and N microanalyses were carried out with CHNS–O Elemental Analyzer Vario EL III, ELEMENTAR, Hanau-Germany while the amount of Fe, W and Cu were measured by inductively coupled plasma mass spectrometry (ICP-MS). Powder XRD was obtained by an X'PertPro Panalytical, Holland diffractometer in 40 kV and 30 mA with a Cu Kα radiation (λ = 1.5418 Å). The XPS spectra of the supported catalyst was recorded on a VG Microtech Twin anode XR3E2 X-ray source and a concentric hemispherical analyzer operated at a base pressure of 5 × 10−10 mbar using Al Kα (hν = 1486.6 eV). Peak fitting of all spectra were undertaken using the Shirley background correction and Gaussian–Lorentzian peak shapes. Binding energies (BEs) were referenced to C 1s peak at 284.5 eV. Low-resolution survey spectra, as well as higher-resolution spectra for C, Fe, O, W, and Cu were collected. Infrared spectra were recorded on a Bruker Vector 22 FT-IR using KBr plate. The morphology of nanocomposites was revealed by a scanning electron microscope (FESEM-TESCAN MIRA3). The elements in the nanocomposite samples were probed with energy-dispersive X-ray (EDX) spectroscopy accessory to the FESEM-TESCAN MIRA3 scanning electron microscopy. The magnetic properties were investigated by a home-made alternative gradient force magnetometer (AGFM) in the magnetic field range of −5000 to 5000 Oe at room temperature. Thermo gravimetric analysis and differential thermal analysis (TGA) were carried out using a STA PT-1000 LINSEIS. PEG with molecular weight of 400 was used in reactions (PEG-400).Preparation of nano materials
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), GO (0.5 g) was added and the mixture sonicated for 30 min. The resulting mixture was deoxygenated by bubbling with nitrogen gas for 10 min, followed by heating to 80 °C for 10 min. NH4OH (10 mL, 8 M) was added rapidly to the heating solution and was left to stir for 1 h. After cooling to ambient temperature, the formed GO–Fe3O4 composites were magnetically collected, washed two times with deionized water (100 mL), and dried at 50 °C.
:2), GO (0.5 g) was added and the mixture sonicated for 30 min. The resulting mixture was deoxygenated by bubbling with nitrogen gas for 10 min, followed by heating to 80 °C for 10 min. NH4OH (10 mL, 8 M) was added rapidly to the heating solution and was left to stir for 1 h. After cooling to ambient temperature, the formed GO–Fe3O4 composites were magnetically collected, washed two times with deionized water (100 mL), and dried at 50 °C.General procedure for the Knoevenagel condensation by HybPOM
HybPOM (0.007 g) was added to a mixture of aromatic aldehyde (1 mmol), malononitrile or ethylcyanoacetate (1.2 mmol) in PEG/water = 1:1 (1 mL), and then the mixture was stirred at room temperature for the appropriate time. The progress was monitored by TLC (eluent: n-hexane/EtOAc, 2:1). After completion of the reaction, 5 mL of EtOAc was added and the catalyst was separated by centrifuge, and then the reaction mixture was extracted with H2O and EtOAc. The organic layer was dried over MgSO4 and then evaporated under reduced pressure. After evaporation of EtOAc, the obtained products were recrystallized from hot EtOH and the pure products were obtained in 85–98% yields (Table 2).
General procedure for the Knoevenagel condensation by GO@Fe3O4@HybPOM
GO@Fe3O4@HybPOM (0.01 g) was added to a mixture of aromatic aldehyde (1 mmol) and malononitrile or ethylcyanoacetate (1.2 mmol) in PEG/water = 1:1 (1 mL), and then the mixture was stirred at room temperature for the appropriate time.
The progress was monitored by TLC (eluent: n-hexane/EtOAc, 2:1). After completion of the reaction, 5 mL of EtOAc was added and catalyst was separated by an external magnet, and then the mixture was extracted with H2O and EtOAc. The organic layer was dried over MgSO4 and then evaporated under reduced pressure. After evaporation of EtOAc, the obtained products were recrystallized from hot EtOH and pure products were obtained in 85–98% yields (Table 2).
General procedure for the synthesis of 2,3-dihydroquinazolin-4(1H)-ones by HybPOM
A mixture of aldehydes (1 mmol), 2-aminobenzamide (1 mmol) and HybPOM (0.005 g) was stirred in ethanol (5 mL) under reflux conditions for the appropriate time, as shown in Table 4. Reaction progress was monitored by TLC. After completion of the reaction (disappearance of starting materials), the reaction mixture was cooled to room temperature, and then the catalyst was separated by centrifuge and reused as such for the next experiment. The decanted was evaporated to remove the solvent; the crude solid product was washed with diethyl ether to obtain pure 2,3-dihydroquinazolin-4(1H)-ones in 90–98% yields (Table 4).General procedure for the synthesis of 2,3-dihydroquinazolin-4(1H)-ones by GO@Fe3O4@HybPOM
A mixture of aldehydes (1 mmol), 2-aminobenzamide (1 mmol) and nanocatalyst (0.007 g) was stirred in ethanol (5 mL) under reflux conditions for the appropriate time as shown in Table 4. Reaction progress was monitored by TLC. After completion of the reaction, the reaction mixture was cooled to room temperature, and then the catalyst was separated by an external magnet and reused as such for the next experiment. The filtrate was evaporated to remove the solvent; the crude solid product was washed with diethyl ether to obtain pure 2,3-dihydroquinazolin-4(1H)-ones in 90–98% yields (Table 4).The NMR spectra data of organic products
For all NMR measurements the chemical shifts are given with respect to tetramethylsilane (TMS) as an internal standard.Results and discussions
In this paper, we report a simple and mild synthesis route through self-assembly polyoxometalate and p-phenylenediamine (PPD) as organic ligand π-electron donor to produce a polyoxometalate-based inorganic–organic hybrid. First, by addition of Cu2+ ion to the lacunary 9-tungstosilicate, β-SiW9O3410− solution, trimetallo derivatives of [SiW9Cu3O37]10− are formed. The solution color is turned from colorless to blue. With addition of PPD molecule to the blue aqueous solution, a blue-brown precipitate formed. PPD molecule via one of its NH2 groups replaced terminal water molecules from substituted Cu2+ ions. The precipitate color finally changed from blue-brown to black because of charge transfer interaction between POM cluster anion and PPD organic molecule.26 The proposed structure for anionic section of nanohybid material is shown in Fig. 1.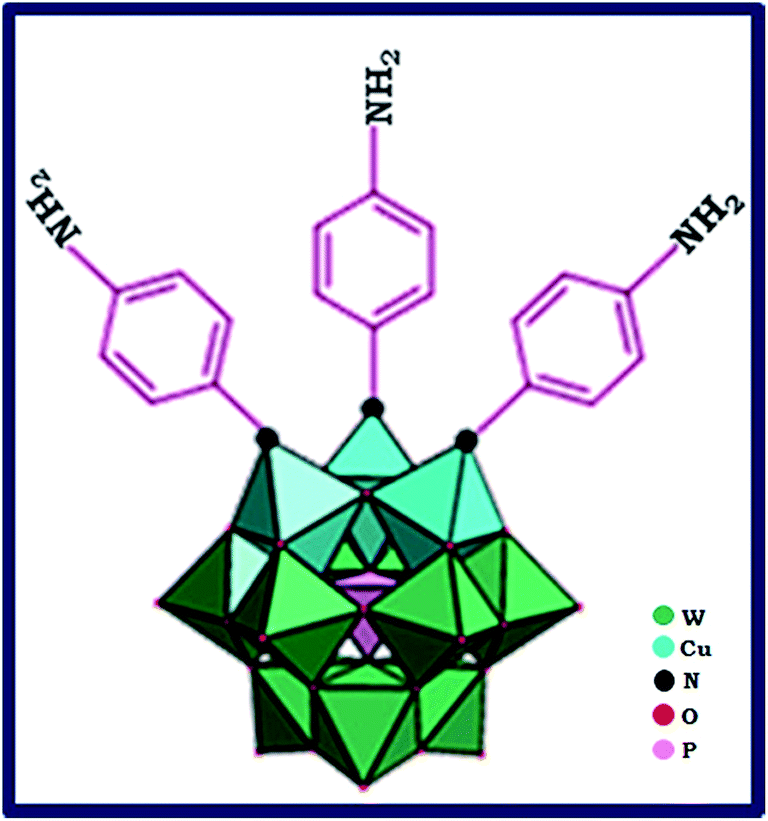 | ||
| Fig. 1 The proposed structure to anionic section of nanohybrid materials of HybPOM. | ||
For easier recovery and separation instead of centrifuge, functionalized nanosized polyoxometalate of HybPOM was placed on the GO–Fe3O4 magnetic nanoparticles. GO–Fe3O4 nanoparticles were prepared by Hummers' method procedure.25
A quantitative analysis of O 1s XPS spectra of GO sample by Gaussian curves fitting show that the relative atomic concentrations (atom%) of carboxyl or carbonyl groups, epoxy, hydroxyl, or carboxyl groups, and oxygen atoms in water and/or chemisorbed oxygen species are 24.08, 47.56 and 28.35%, respectively.
Due to abundant functional groups (such as epoxy, hydroxyl, and carboxyl groups) on the surface, graphene oxide (GO) is ready to the attack the nucleophiles.
Organic compounds with functional amine groups may be loaded onto GO surface through the nucleophilic substitution followed by formation of C–N bond. It may also be loaded onto the surface of graphene oxide by the chemical reaction between amino groups and carboxylic acid groups on the edge of GO sheets (Scheme 1). Such methods are strong enough to firmly deposit organic substrates with amine groups on the graphene oxide sheets.27
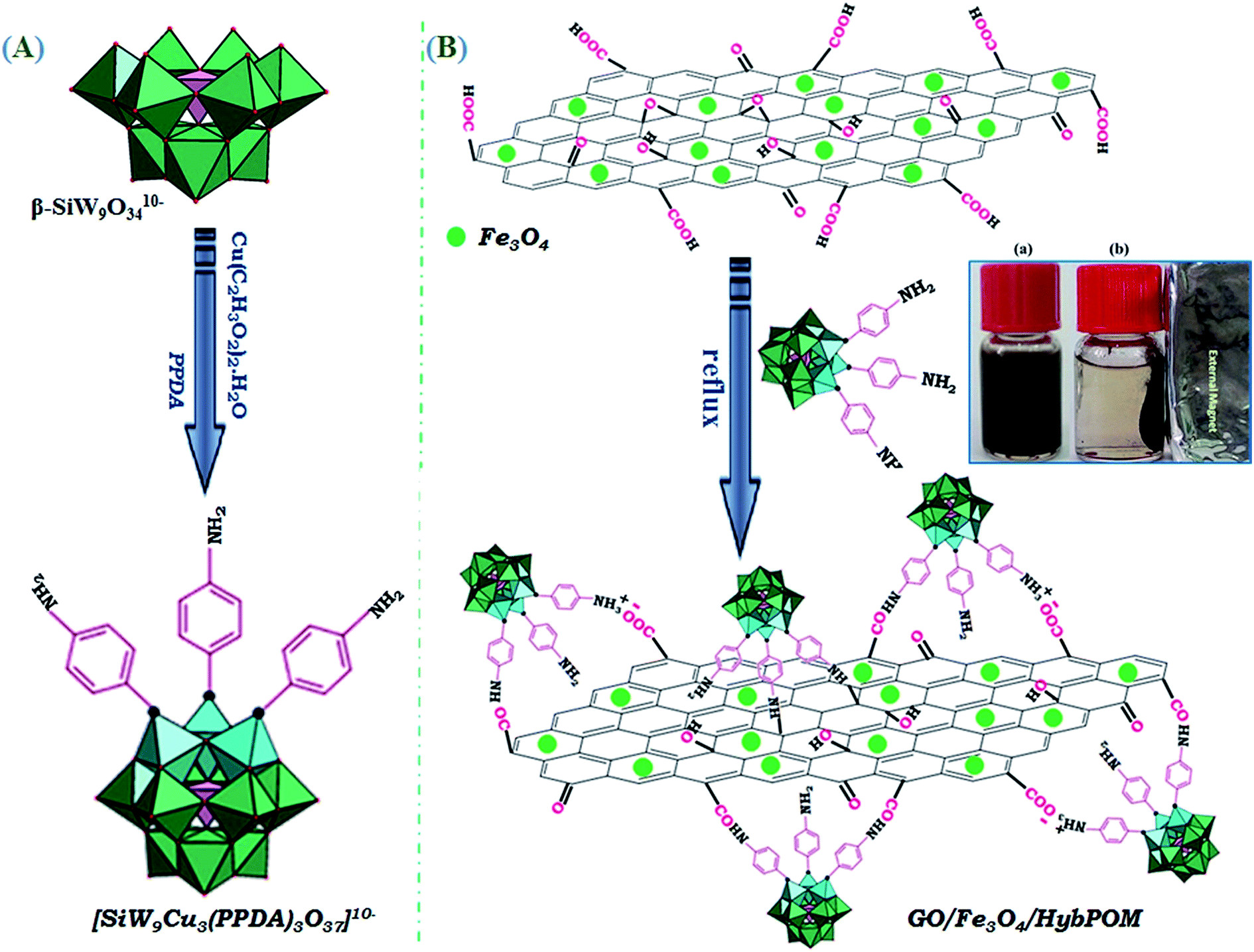 | ||
| Scheme 1 Preparation of the nanohybrid of HybPOM, and magnetically-recoverable nanocomposite of GO@Fe3O4@HybPOM. | ||
Common methods such as titration with Hammett indicators and temperature programmed desorption of adsorbed molecules such as ammonia or pyridine, adsorption microcalorimetry and NMR spectroscopy have been employed to describe the acidity of POMs in the solid state both qualitatively and quantitatively. Potentiometric titration with n-butylamine allows us to estimate the number of acid sites and their distribution.28 The titration curves show that HybPOM and Go/Fe3O4/HybPOM with the initial electrode potential −165.9 and −73.8 mV are classified as very weak and weak acids, respectively. Difference in the acid strength of HybPOM and Go/Fe3O4/HybPOM is attributed to the total number of free NH2 groups on them. For Go/Fe3O4/HybPOM, some of the NH2 groups are involved in the bonding with Go/Fe3O4 nanoparticle. The titration curves of HybPOM and Go/Fe3O4/HybPOM is presented in the ESI (Fig. S3†).
FT-IR spectra of HybPOM hybrid material show the characteristic band of PPD and the trimetallo derivatives of [SiW9O37Cu3(H2O)3]10− heteropolyanions moieties with some variations in the band vibrational frequencies or intensities (Fig. 2). The alkaline salts of complex anions which often crystallize with many water molecules and involve hydrogen bonds lead to a decrease in the frequencies of the metal–oxygen stretching. With large organic PPD molecules,29 as is expected, the effects arising from cation size, cation polarizing power and crystallization solvent molecules are diminished so that the POM characteristic bands are observed clearly in the range 1000–700 cm−1.
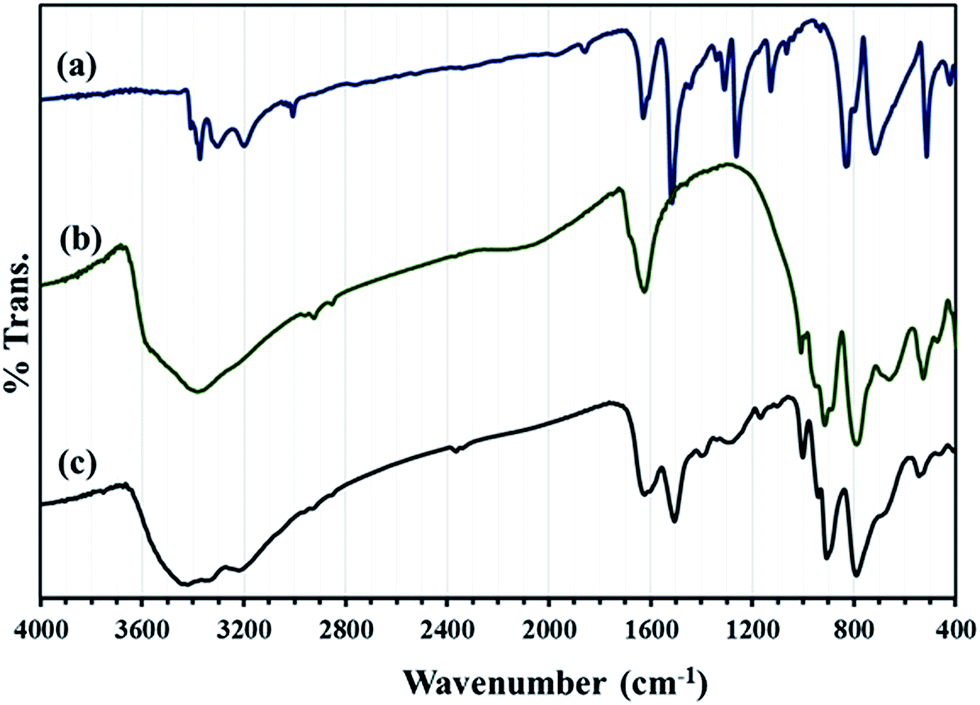 | ||
| Fig. 2 FT-IR spectra of (a) PPD, (b) [SiW9O37Cu3(H2O)3]10− and (c) HybPOM. | ||
The N–H stretch band (3300–3400 cm−1) from PPD molecule overlap with OH stretching vibration band (∼3500 cm−1) from water molecule but other bands can be observed in the IR spectra of HybPOM hybrid materials.
In Fig. 3 FT-IR spectra of GO, GO/Fe3O4, HybPOM were given for comparison with that of GO@Fe3O4@HybPOM.
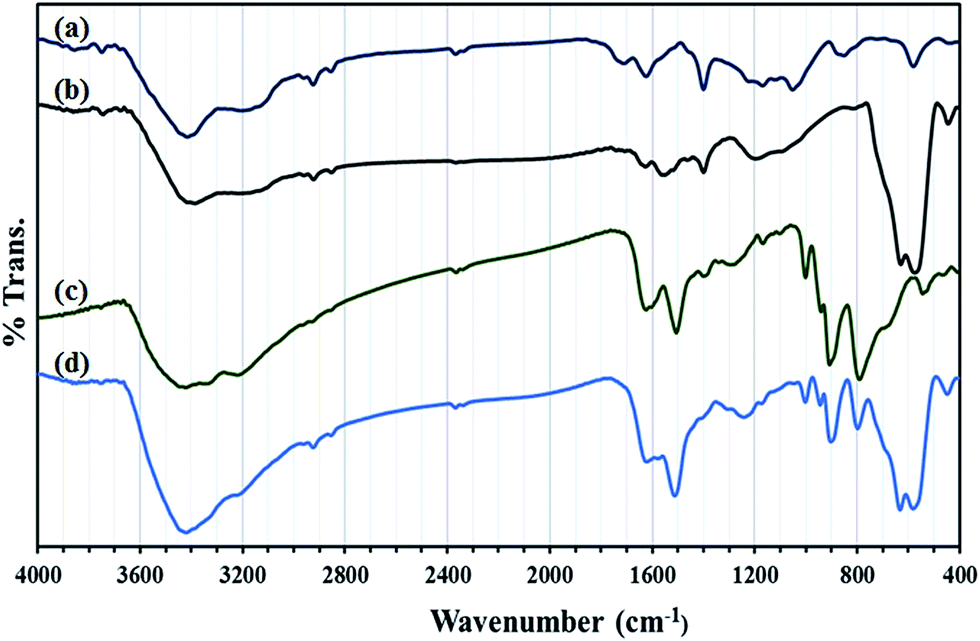 | ||
| Fig. 3 FT-IR spectra of (a) GO, (b) GO/Fe3O4, (c) HybPOM and (d) GO@Fe3O4@HybPOM. | ||
Peaks at 573–630 cm−1 in the IR spectra of GO/Fe3O4 and GO@Fe3O4@HybPOM are attributed to Fe–O stretching vibration. The complete IR pattern of corresponding trimetallo POM of [SiW9Cu3O37]10− with some variations in band frequencies and intensities were observed in those of GO@Fe3O4@HybPOM in range of 1000–700 cm−1. A small blue shift is observed in the frequencies of the band stretching due to disappearance of anion–anion interactions. The vibration modes of the quinoidal benzene rings appear at 1510 and 1620 cm−1 for GO@Fe3O4@HybPOM which overlapped with the bending vibrations of amide bond of N–H.30 The C–N stretching vibrations appear at 1200–1400 cm−1. The O–H and N–H stretching vibrations appear as broad and strong bands at range of 3200–3650 cm−1.
The GO–Fe3O4 powder exhibit a total mass loss of 12.35% at 100–800 °C, which are attributed to the removal of adsorbed water and the decomposition and vaporization of various oxygen-containing functional groups at different positions on the surface of the GO–Fe3O4 (Fig. 4a).
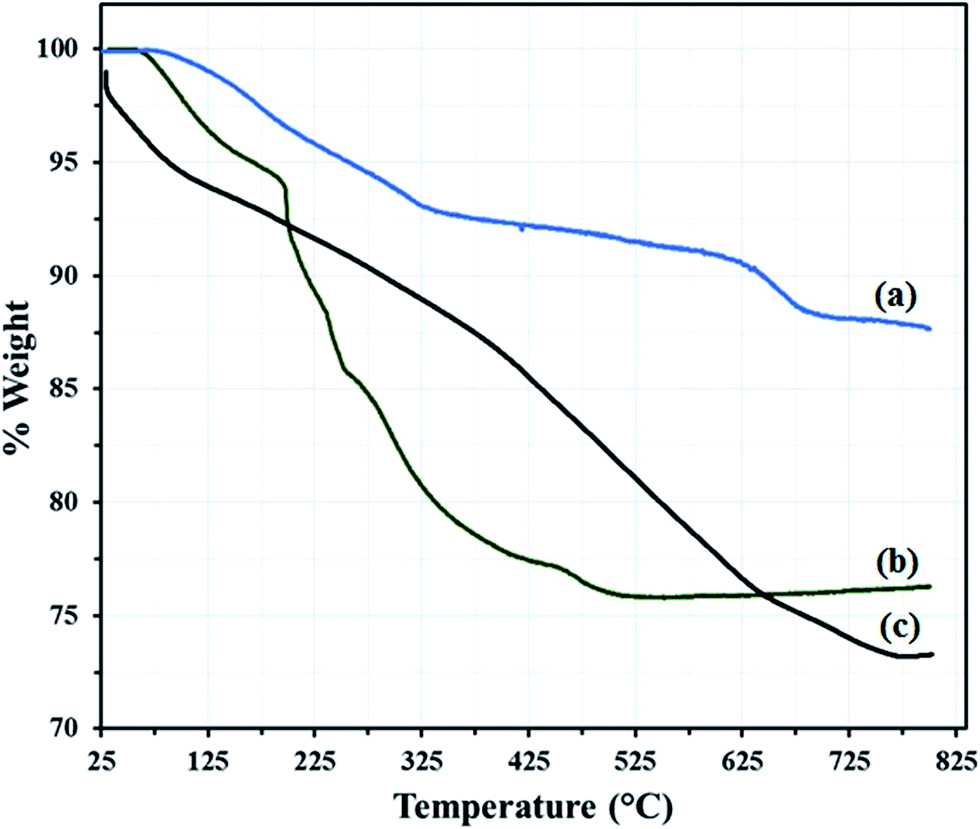 | ||
| Fig. 4 TG analysis of (a) GO@Fe3O4, (b) HybPOM and (c) GO@Fe3O4@HybPOM. | ||
The thermogravimetric analysis (TGA) curve of the hybrid materials HybPOM show two mass loss regions below 600 °C (Fig. 4b). First, 30–200 °C with 6.12 weight loss which are assigned to lattice water loss equal to 12 (6.24%) molecules for HybPOM. Second, 200–550 °C with approximately 18.19 weight loss for HybPOM is attributed to loss of 6 (18.73%) PPD molecules.
Results of TGA are confirmed well by CHN analysis of HybPOM. TGA of GO@Fe3O4@HybPOM (Fig. 4c) nanocomposite exhibit two steps of mass loss at 25–225 °C and 225–800 °C with 8.60 and 17.50% weight loss, respectively, which are attributed to the release of water and PPD molecules along oxygen-containing functional groups on GO–Fe3O4 surface.
This large weight loss indicates the loading of the polyoxometalate-based inorganic–organic hybrid material on the GO–Fe3O4. From the TGA curves, it can be concluded that GO@Fe3O4@HybPOM nanocomposites exhibit a higher thermal stability than HybPOM nanohybrides. This enhancement in the thermal stability is due to some ionic interaction between graphene–Fe3O4 and the amine group of the aniline ring. These may form a coordinate bond between Fe and N, as Fe can facilitate back bonding between the d-orbital and NH2 group of the aniline ring.30
The compounds of β-SiW9O3410− (Fig. 5a) and PDA are crystalline in the solid state, while X-ray powder diffractions show that HybPOM is typical amorphous state, implying that the hybrid materials have less-ordered structures (Fig. 5b). However, it can be observed that the XRD of HybPOM contain XRD pattern of both β-SiW9O3410− and PPD. X-ray diffraction (XRD) measurements were also performed to obtain crystalline structural information for the GO, GO–Fe3O4, GO@Fe3O4@HybPOM. The broad diffraction peak around 18.72° corresponds to C (002) reflection of GO (Fig. 5 inset).30
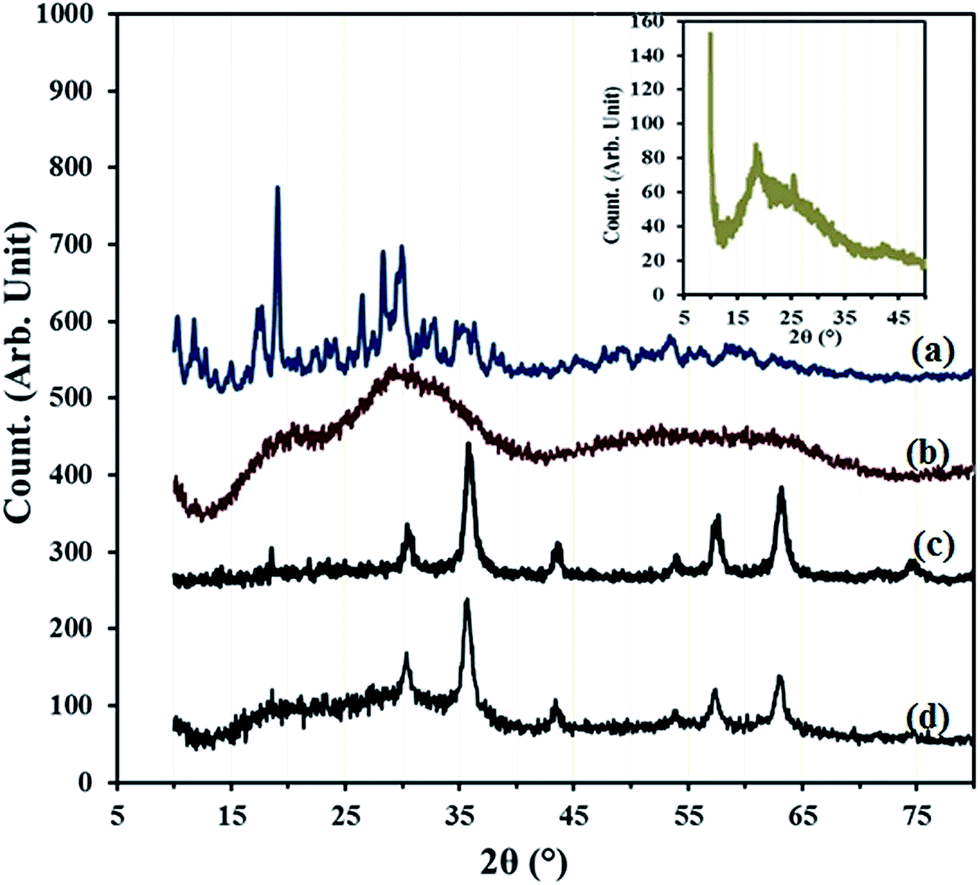 | ||
| Fig. 5 XRD spectra of (a) Na9H[β-XW9O34]·nH2O, (b) HybPOM, (c) GO@Fe3O4 (d) GO@Fe3O4@HybPOM and (inset) GO. | ||
At the GO–Fe3O4, the intense diffraction peaks indexed to (220), (311), (400), (422), (511), (440), and (533) planes appearing at 2θ = 30.52°, 35.85°, 43.58°, 53.96°, 57.43°, 63.16° and 74.65°,respectively, and the peak positions and relative intensities match well with the standard XRD data for the cubic phase Fe3O4 with a face-centered cubic (fcc) structure (JCPDS no. 19-629) (Fig. 5c).31 Disappearance of the reflection plane at (002) and merging of the planes of Fe3O4 show the good interfacial interaction between the planes. The XRD of GO@Fe3O4@HybPOM show corresponding peaks of the GO–Fe3O4 and the amorphous XRD pattern of HybPOM as background (Fig. 5d).
The SEM images of HybPOM reveal the relatively uniform spherical nanometer particles with a diameter of less than 80 nm (Fig. 6a). The SEM images of GO sheets, GO–Fe3O4, and GO@Fe3O4@HybPOM composites are presented in Fig. 6b and c. The images illustrate that the GO sheets shows a stacked and crumpled morphology (Fig. 6b).32 From the SEM image, it could be observed that the surface of GO sheets were decorated tightly with Fe3O4 nanoparticles (Fig. 6c).
 | ||
| Fig. 6 SEM images of (a) HybPOM, (b) GO, (c) GO@Fe3O4 (d) GO@Fe3O4@HybPOM. | ||
Interestingly, Fe3O4 nanoparticles were only observed on the basal surface and the edges of GO at very high coverage and without significant aggregation. This indicates that Fe3O4 nanoparticles were mainly nucleated and grown on GO substrate and that their free nucleation was effectively suppressed. The stacking of GO–Fe3O4 observed in SEM images is probably due to strong magnetic interaction among Fe3O4 nanoparticles in neighboring layers. The SEM images of GO@Fe3O4@HybPOM show that these magnetic nanocomposites are uniform with a mean size of ∼50 nm particles, but with some self-aggregation (Fig. 6d).
In order to shed more light on the elemental composition and preparation progress, the chemical compositions of HybPOM and GO@Fe3O4@HybPOM were analyzed by EDX. The EDX analysis of HybPOM hybrid material shows peaks of W, O, Si, C, N, Cu (Fig. 7a).
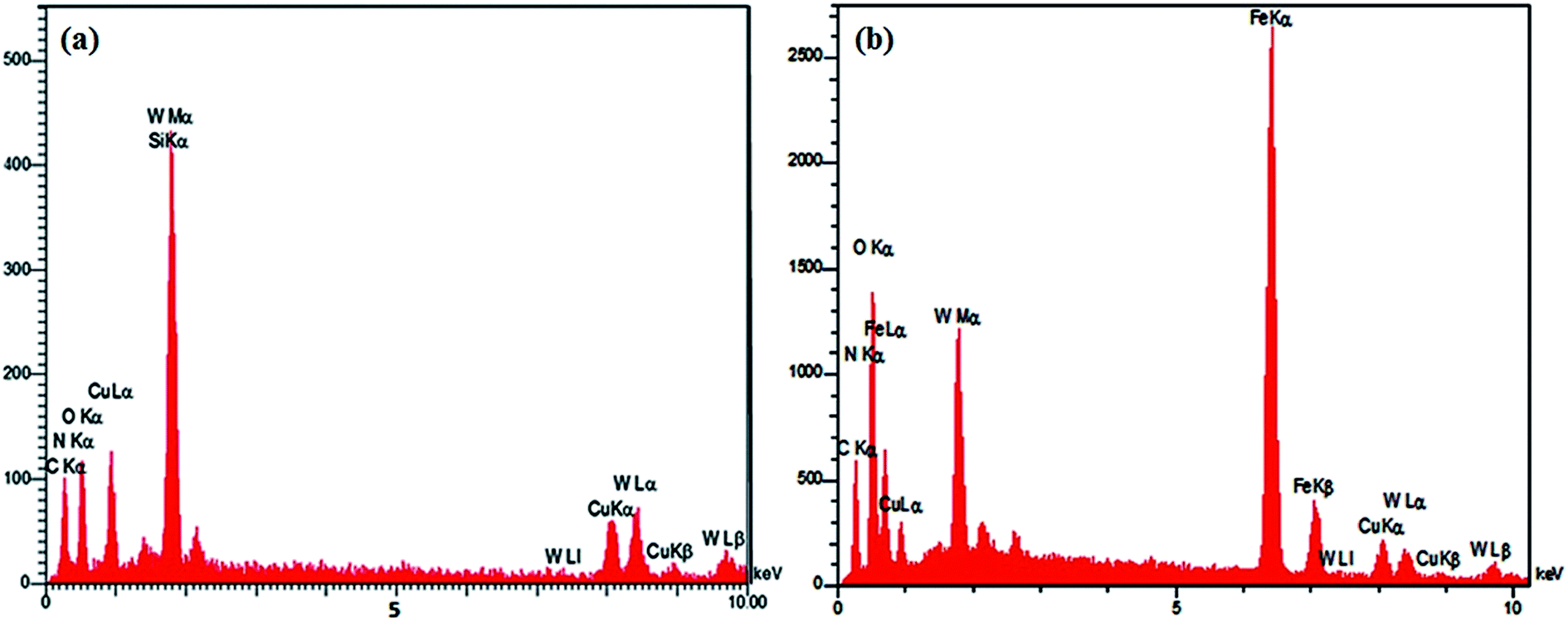 | ||
| Fig. 7 EDX spectra of (a) HybPOM, (b) GO@Fe3O4@HybPOM. | ||
The magnetization of GO–Fe3O4 and GO@Fe3O4@HybPOM was measured at room temperature, as shown in Fig. 8. The magnetic hysteresis loops are S-like curves. For GO–Fe3O4 hybrid, the magnetic remanence was nearly zero (Fig. 8a). This indicated that there was almost no remaining magnetization when the external magnetic field was removed, suggesting that GO–Fe3O4 exhibit a superpara magnetic behavior.
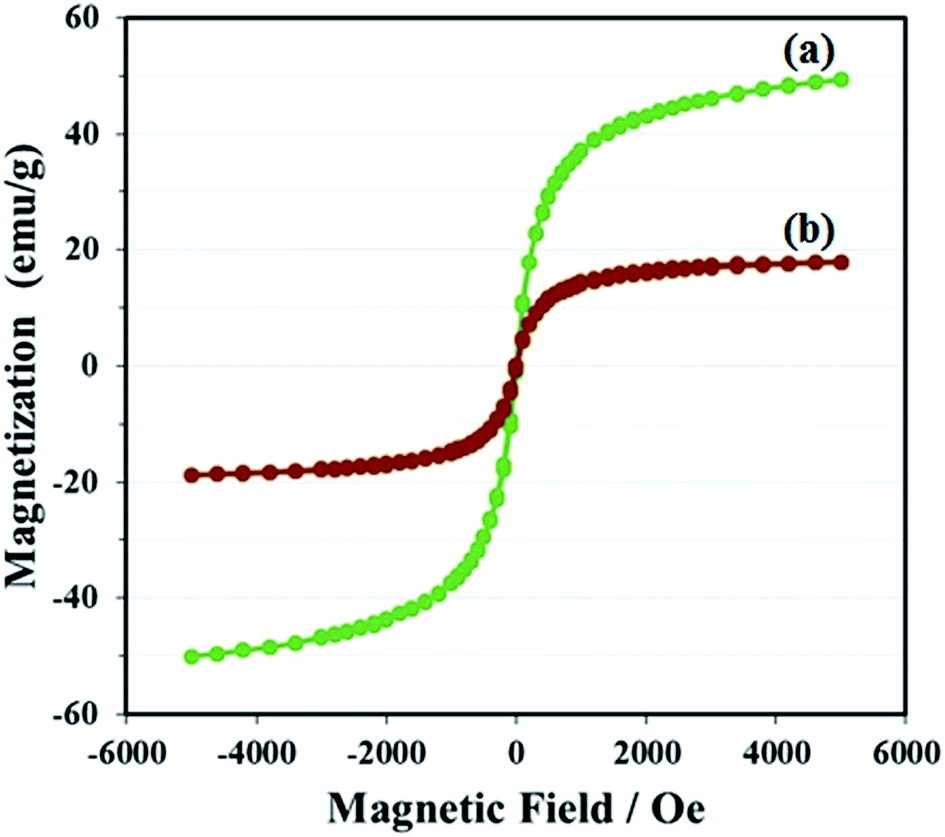 | ||
| Fig. 8 Magnetization curves of (a) GO@Fe3O4, (b) GO@Fe3O4@HybPOM. | ||
The specific saturation magnetization of GO–Fe3O4, is 49.31 emu g−1, which is smaller than the reported value of bulk Fe3O4 of 92 emu g−1.33 The specific saturation magnetization of GO@Fe3O4@HybPOM is 17.94 emu g−1 (Fig. 8b). Decrease in saturation magnetization observed for GO@Fe3O4@HybPOM could be attributed to the increased mass and the loading polyoxometalate-based inorganic–organic hybrids. Even with this reduction in the saturation magnetization, complete magnetic separation of GO@Fe3O4@HybPOM was achieved in <10 s by placing a magnet near the vessels containing the aqueous dispersion of the nanoparticles.
XPS analysis was used to gain further insights into chemical compositions of samples. Fig. 9a depicts the XPS survey spectrum of GO@Fe3O4@HybPOM. As expected, W 4f, C 1s, N 1s, O 1s, Fe 2p and Cu 2p peaks were observed in the XPS survey spectrum. The W 4f spectrum can be deconvoluted into doublets, as indicated in Fig. 9b. This doublet consists of W 4f7/2 line at 35.2 eV and W 4f5/2 line at 37.2 eV, which are assigned to the W in the W–O bond configuration and typically observed for the W6+.34
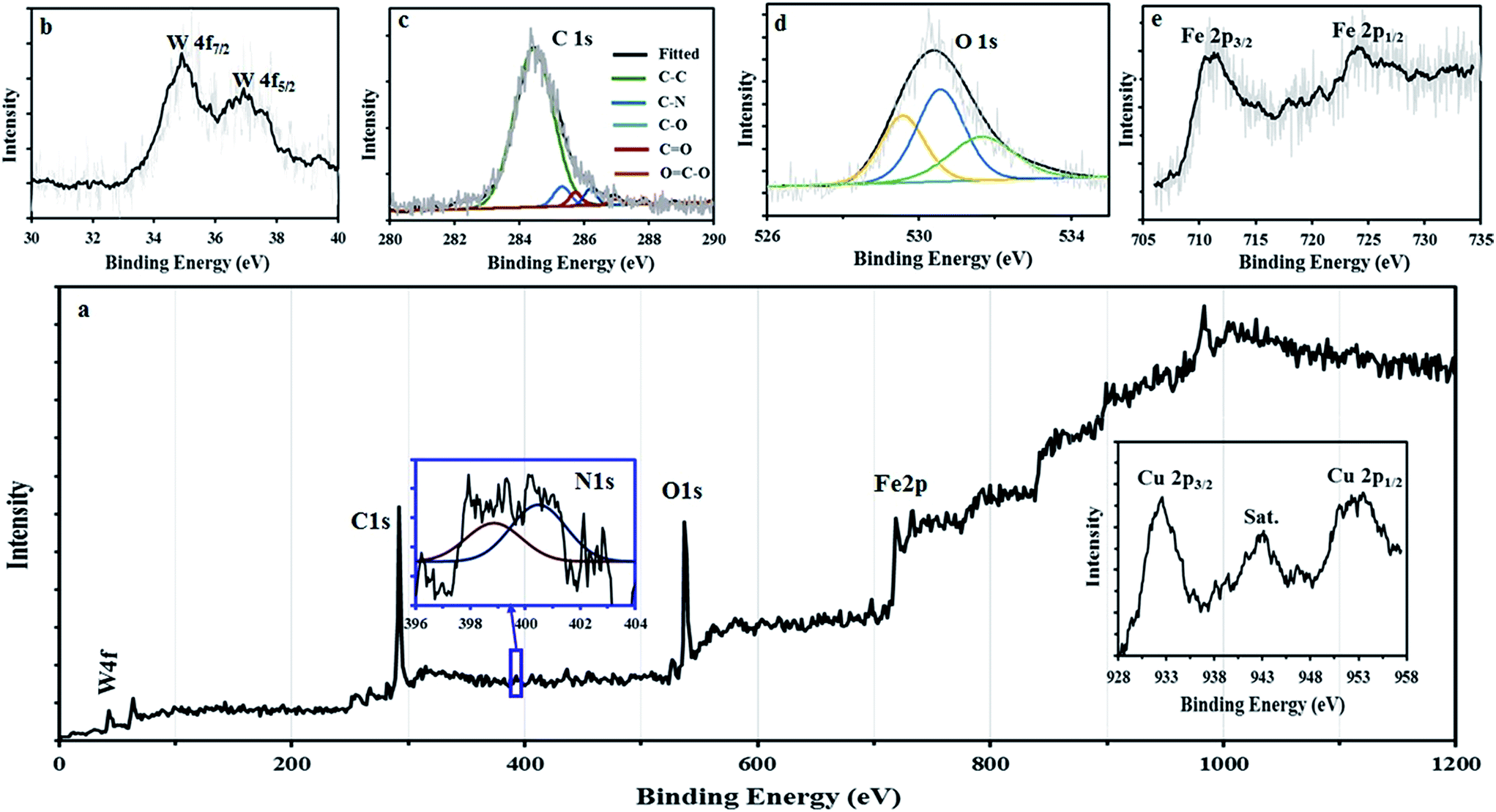 | ||
| Fig. 9 XPS survey spectra of (a) GO@Fe3O4@HybPOM and high-resolution XPS spectra of N 1s (inset), and Cu 2p (inset) (b) W 4f, (c) C 1s, (d) O 1s, (e) Fe 2p. | ||
The peaks observed at 284.5 and 530.4 eV (Fig. 9c and d) can be assigned to the binding energy of C 1s and O 1s, respectively. From the C 1s XPS spectrum, different peaks corresponding to C–C, C–N, C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O are observed at 284.5, 285.3, 286.0 and 286.8, respectively. The peak of the COOH at 287.8 eV almost disappeared upon the amide formation with the amine groups of HybPOM. The binding energy appears at 399–402 eV which could be attributed to the N 1s protonated and nonprotonated N atoms (Fig. 9a inset). The binding energy peaks at 711.8 and 725.1 eV (Fig. 9e) are corresponding to Fe 2p3/2 and Fe 2p1/2, respectively, which are very close to the literature values.35 The disappearance of charge transfer satellite of Fe 2p3/2 around 720 eV reveals the formation of mixed oxides of Fe2+ and Fe3+ such as Fe3O4. The XPS spectra from 953 to 935 eV demonstrate the photoelectron spectrum of the Cu 2p. As shown in Fig. 9a (inset), there are two main Cu 2p XPS peaks at 933.2 and 953.1 eV, which could be due to the Cu2+ double peaks for Cu 2p3/2 and Cu 2p1/2, respectively.36–38 The satellite peak at 941.6 eV located at higher binding energy than that of the main Cu 2p3/2 peak, is typically associated with copper in the bivalent binding energy state.36–39
O are observed at 284.5, 285.3, 286.0 and 286.8, respectively. The peak of the COOH at 287.8 eV almost disappeared upon the amide formation with the amine groups of HybPOM. The binding energy appears at 399–402 eV which could be attributed to the N 1s protonated and nonprotonated N atoms (Fig. 9a inset). The binding energy peaks at 711.8 and 725.1 eV (Fig. 9e) are corresponding to Fe 2p3/2 and Fe 2p1/2, respectively, which are very close to the literature values.35 The disappearance of charge transfer satellite of Fe 2p3/2 around 720 eV reveals the formation of mixed oxides of Fe2+ and Fe3+ such as Fe3O4. The XPS spectra from 953 to 935 eV demonstrate the photoelectron spectrum of the Cu 2p. As shown in Fig. 9a (inset), there are two main Cu 2p XPS peaks at 933.2 and 953.1 eV, which could be due to the Cu2+ double peaks for Cu 2p3/2 and Cu 2p1/2, respectively.36–38 The satellite peak at 941.6 eV located at higher binding energy than that of the main Cu 2p3/2 peak, is typically associated with copper in the bivalent binding energy state.36–39
Catalytic studies
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | HybPOM (GO@Fe3O4@HybPOM) (g) | Solvent | T (°C) | HybPOM | GO@Fe3O4@HybPOM | ||||
| Time | Conversion | Yieldb (%) | Time | Conversion | Yieldb (%) | ||||
| a Reaction conditions: benzaldehyde (1 mmol), malononitrile (1.2 mmol), solvent (1 mL), r.t, air.b Isolated yield.c The bold letters represent the most effective reaction conditions. | |||||||||
| 1 | 0(0) | H2O | r.t | 10 h | Trace | Trace | 10 h | Trace | Trace |
| 2 | 0.02(0.02) | H2O | r.t | 10 h | 30 | 20 | 10 h | 40 | 35 |
| 3 | 0.04(0.04) | H2O | 70 | 10 h | 50 | 45 | 10 h | 40 | 37 |
| 4 | 0.005(0.007) | PEG/H2O | r.t | 70 min | 100 | 80 | 60 min | 100 | 98 |
| 5c | 0.007(0.010) | PEG/H2O | r.t | 10 min | 100 | 96 | 20 min | 100 | 98 |
| 6 | 0.010(0.01) | PEG/H2O | r.t | 10 min | 100 | 95 | 20 min | 100 | 96 |
| 7 | 0.030(0.03) | PEG/H2O | r.t | 10 min | 100 | 94 | 20 min | 100 | 95 |
| 8 | 0.007(0.010) | PEG/H2O | 70 | 5 min | 100 | 95 | 10 h | 100 | 97 |
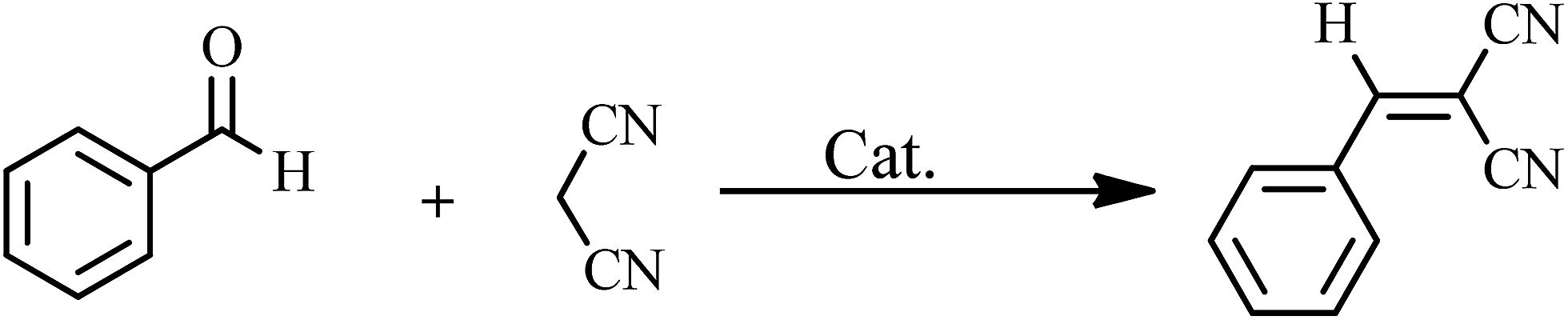
As shown in Table 1, the amount of 0.007 g of HybPOM was found to be ideal for the Knoevenagel condensations and the best results were obtained using PEG/water = 1:1 (PEG, polyethylene glycol) as solvent. A series of aromatic aldehydes carrying either electron-donating or electron-withdrawing substitutions were subjected to the optimized reaction conditions and the expected products were obtained in short time periods and at high yields (Table 2).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | HybPOM (0.007 g) | GO@Fe3O4@HybPOM (0.01 g) | Conversion (%) | Mp (°C) [Ref.] | ||
| Time (min) | Yieldb,c (%) | Time (min) | Yieldb,c (%) | |||||
| a Reaction conditions: aromatic aldehyde (1 mmol), malononitrile or ethylcyanoacetate (1.2 mmol), PEG/H2O (1 mL), r.t, air.b Isolated yield.c All the products were identified and characterized by comparison of their physical and spectral data with those reported in the literature (see ESI). | ||||||||
| 1 | C6H4 | CN | 10 | 96 | 20 | 98 | 100 | 80–82(82–83)55 |
| 2 | 4-CH3OC6H4 | CN | 45 | 95 | 50 | 95 | 100 | 111–113(112–114)55 |
| 3 | 3-NO2C6H4 | CN | 15 | 90 | 15 | 97 | 100 | 99–101(101–103)56 |
| 4 | 4-NO2C6H4 | CN | 15 | 96 | 20 | 97 | 100 | 156–158(158–159)55 |
| 5 | 2-ClC6H4 | CN | 35 | 95 | 70 | 93 | 100 | 80–82(79–82)57 |
| 6 | 4-ClC6H4 | CN | 20 | 89 | 30 | 94 | 100 | 162–163(162–164)58 |
| 7 | 4-CH3OC6H4 | CO2Et | 125 | 85 | 130 | 85 | 100 | 78–80(79–81)55 |
| 8 | 3-NO2C6H4 | CO2Et | 25 | 85 | 35 | 95 | 100 | 130–131(130–132)59 |
| 9 | C6H4 | CO2Et | 60 | 90 | 70 | 95 | 100 | 49–51(50–52)55 |
| 10 | 4-NO2C6H4 | CO2Et | 20 | 95 | 30 | 97 | 100 | 169–170(169–170)60 |
| 11 | 2-ClC6H4 | CO2Et | 130 | 95 | 140 | 92 | 100 | 48–50(48–50)61 |
| 12 | 4-ClC6H4 | CO2Et | 140 | 93 | 150 | 88 | 100 | 86–88(87–89)60 |
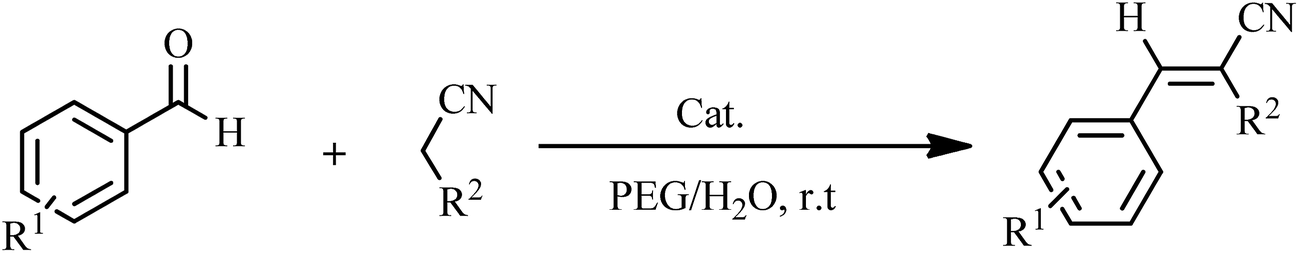
There are several advantages for supporting HybPOM on the GO–Fe3O4, such as, easier recovery, improvement of efficiency and magnetic separation. Knoevenagel condensation was carried out to investigative the catalytic activity of GO@Fe3O4@HybPOM nanocomposites.
As shown in Table 1, amount of 0.010 g of GO@Fe3O4@HybPOM was found to be ideal for the Knoevenagel condensations and the best results were obtained using PEG/water = 1:1 (PEG, polyethylene glycol) as solvent.
Like the above conditions, Knoevenagel condensation was probed by GO@Fe3O4@HybPOM catalysts under the optimized reaction conditions (Table 2). As shown in Table 1, the expected products were obtained in short time periods and at high yields.
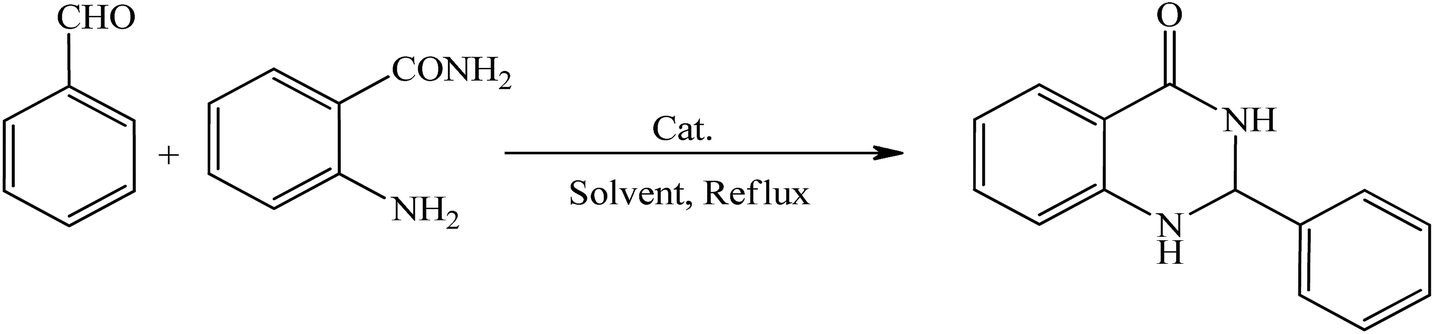
Several strategies for their synthesis were already developed: condensation of anthranilamide with an aldehyde or ketone using p-toluenesulfonic acid as a catalyst,45 desulfurization of 2-thioxo-4(3H)-quinazolinones,46 reaction of isatoic anhydride with Schiff-bases,47 one-step conversion of 2-nitrobenzamides to 2,3-dihydro-4(1H)-quinazolinones,48 and a one-pot three-component condensation of isatoic anhydride, aldehydes and amines.49
The catalysts reported for the above stated one-pot procedure include inorganic catalysts like aluminiumtris (dihydrogen phosphate),50 silica sulfuric acid,51 magnetic Fe3O4 particles,52 reaction media like ionic liquid53 and Asymmetric Brønsted acid.54 Some of these methodologies involve strongly acidic conditions, toxic catalysts, hazardous organic solvents and long reaction times. Thus, there is a need for a non-acidic and novel catalyst that could overcome the above drawbacks.
After successfully synthesizing a series of benzylidenemalononitrile derivatives in excellent yields, we also explored the use of this catalyst system for the synthesis of 2,3-dihydroquinazolin-4(1H)-ones from aldehyde derivatives and 2-aminobenzamide.
The preliminary investigation was to screen the solvent and different amounts of catalyst on the reaction of benzaldehyde with 2-aminobenzamide (Table 3). The model reaction was carried out in the presence of different solvents, temperatures and amounts of catalyst. The best results were obtained with 2-aminobenzamide (1 mmol) and benzaldehyde (1 mmol) in the presence of catalyst HybPOM (0.005 g) and EtOH (5 mL) as solvent under reflux condition (Table 3, entry 3).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | HybPOM (GO@F3O4@HybPOM) (g) | Solvent (1:1) |
HybPOM | GO@F3O4@HybPOM | ||||
| Time (min) | Conversion (%) | Yielda (%) | Time (min) | Conversion (%) | Yielda (%) | |||
| a Isolated yield.b The bold letters represent the most effective reaction conditions.c Solution atrt.d Solution at 100 °C.e Solution at 100 °C. | ||||||||
| 1 | 0(0) | EtOH | 200 | Trace | Trace | 200 | Trace | Trace |
| 2 | 0.003(0.003) | EtOH | 60 | 85 | 80 | 70 | 80 | 70 |
| 3b | 0.005(0.007) | EtOH | 25 | 100 | 96 | 45 | 100 | 85 |
| 4 | 0.007(0.10) | EtOH | 25 | 100 | 97 | 30 | 100 | 97 |
| 5c | 0.005(0.007) | EtOH | 150 | 65 | 60 | 30 | 73 | 95 |
| 6d | 0.005(0.007) | PEG/H2O | 35 | 75 | 70 | 150 | 75 | 70 |
| 7e | 0.005(0.007) | PEG | 30 | 77 | 75 | 25 | 82 | 80 |
| 8 | 0.005(0.007) | EtOAc | 60 | 75 | 70 | 70 | 77 | 75 |
| 9 | 0.005(0.007) | CH2Cl2 | 110 | 75 | 75 | 90 | 87 | 85 |
After optimizing the reaction conditions, the reaction of various aldehydes with 2-aminobenzamide were tested. Results are summarized in Table 4. Synthesis of 2,3-dihydroquinazolin-4(1H)-one derivatives was carried out to investigative catalytic activity of GO@Fe3O4@HybPOM nanocomposite.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Aldehyde | HybPOM (0.005 g) | GO@Fe3O4@HybPOM (0.007) | Mp (°C) [Ref.] | ||
| Time (min) | Yielda,b (%) | Time (min) | Yielda,b (%) | |||
| a All the products were identified and characterized by comparison of their physical and spectral data with those reported in the literature (see ESI).b Isolated yield. | ||||||
| 1 |  |
30 | 97 | 30 | 97 | 223–224(224–226)62 |
| 2 |  |
30 | 90 | 40 | 98 | 197–200(198–200)52 |
| 3 |  |
45 | 88 | 50 | 93 | 184-185(184)54 |
| 4 |  |
40 | 95 | 45 | 95 | 202–203(198–201)62 |
| 5 | 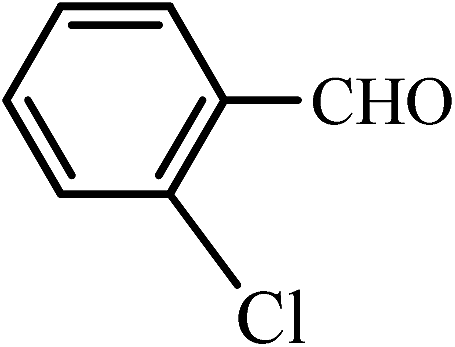 |
55 | 92 | 55 | 90 | 228–230(230–231)63 |
| 6 | 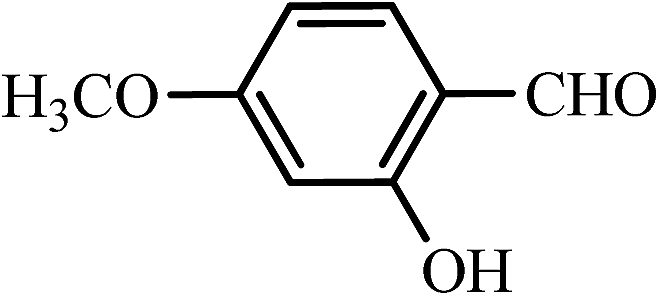 |
30 | 95 | 30 | 96 | 262–263(262–263)64 |
| 7 | 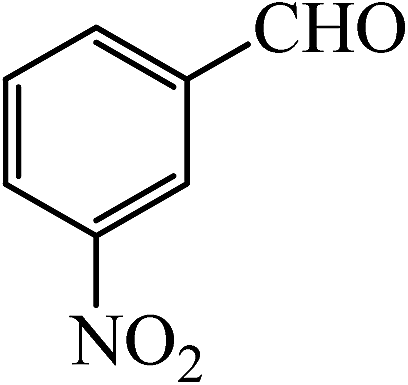 |
50 | 90 | 60 | 90 | 180–182(180–182)63 |
| 8 | 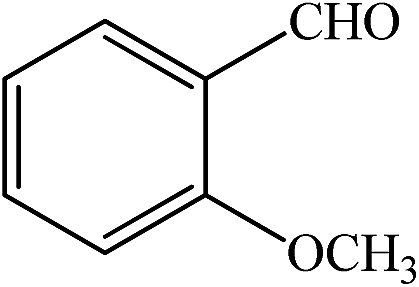 |
35 | 92 | 45 | 95 | 165–167(165–166)65 |
| 9 | 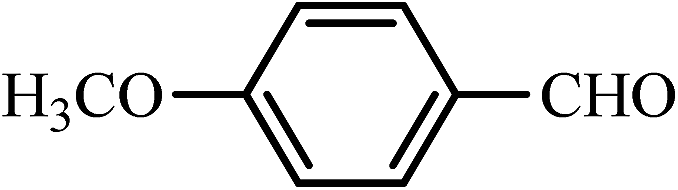 |
25 | 90 | 30 | 95 | 182–183(182–183)64 |
| 10 | 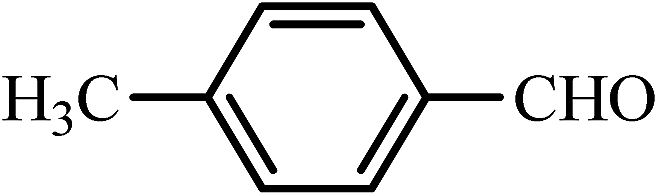 |
40 | 93 | 50 | 95 | 225–226(223–225)49 |
The model reaction was carried out in the presence of different solvents, various temperatures and different amounts of catalyst. The best results were obtained with 2-aminobenzamide (1 mmol) and benzaldehyde (1 mmol) in the presence of catalyst GO@Fe3O4@HybPOM (0.007 g) and EtOH (5 mL) as solvent under reflux conditions (Table 3, entry 3). After optimizing the reaction conditions, the reaction of various aldehydes with 2-aminobenzamide were tested. Results are summarized in Table 4.
Catalysts recovery and reuse
Finally, the reusability of the nanohybrid and nanocomposite were investigated. Fig. 10 shows the yield of five consecutive cycles for the preparation of 2-benzylidenemalononitrile (a) and 2-phenyl-2,3-dihydroquinazolin-4(1H)-one (b) by nanohybrid of HybPOM and GO@Fe3O4@HybPOM, respectively.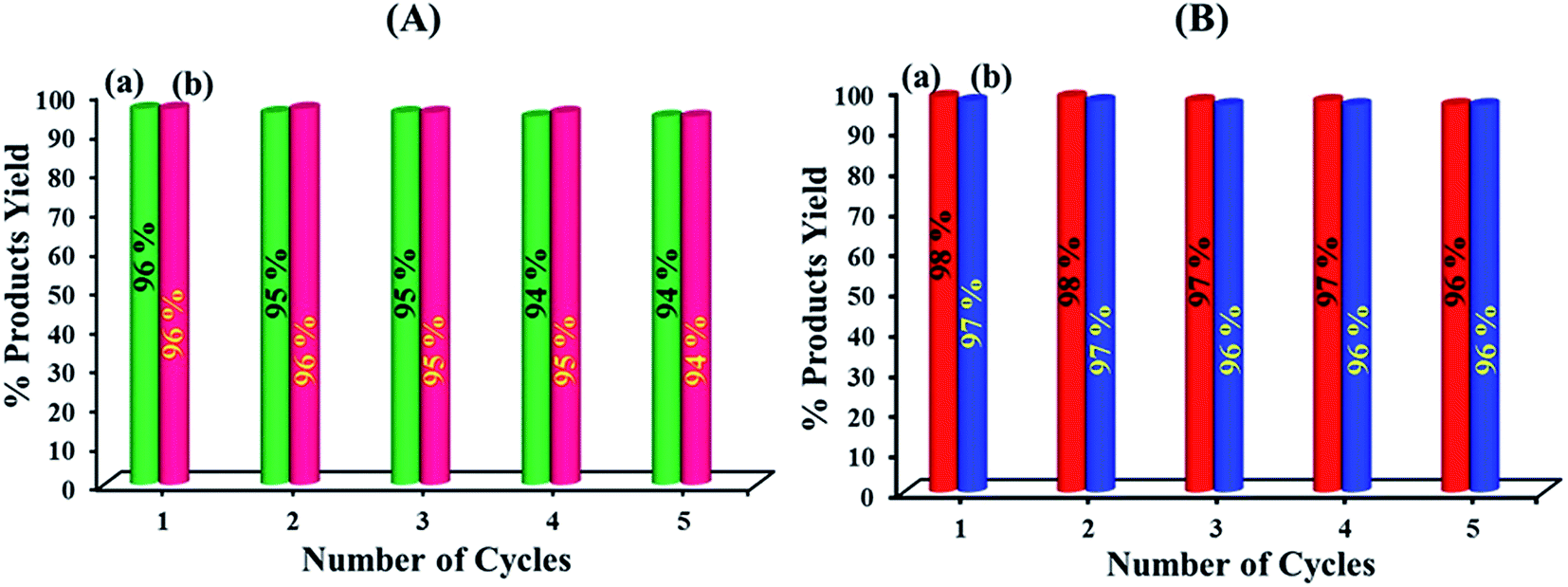 | ||
| Fig. 10 Reusability of HybPOM (A) and GO@Fe3O4@HybPOM (B) in the reaction of benzaldehyde with malononitrile (columns a) and the reaction of 2-aminobenzamide with benzaldehyde (columns b). | ||
After the reaction reached completion, the catalyst was recovered, washed with EtOAc and then reused in the next run. The heterogeneous catalyst showed good recyclability and can be reused for at least five times without significant loss of its catalytic activity. The FT-IR spectrum showed no significant structural changes for catalysts after five consecutive runs (Fig. 11). In order to determine whether the metal leaches out from the solid catalyst during reaction, the hot filtration test was undertaken for the synthesis of 2-(3-bromophenyl)-2,3-dihydoquinazolin-4(1H)-one from the reaction of 2-aminobenzamide with 3-bromobenzaldehyde. The catalyst was separated by applying an external magnet when the reaction had proceeded to nearly 50% completion, and the filtrate was allowed to react further.
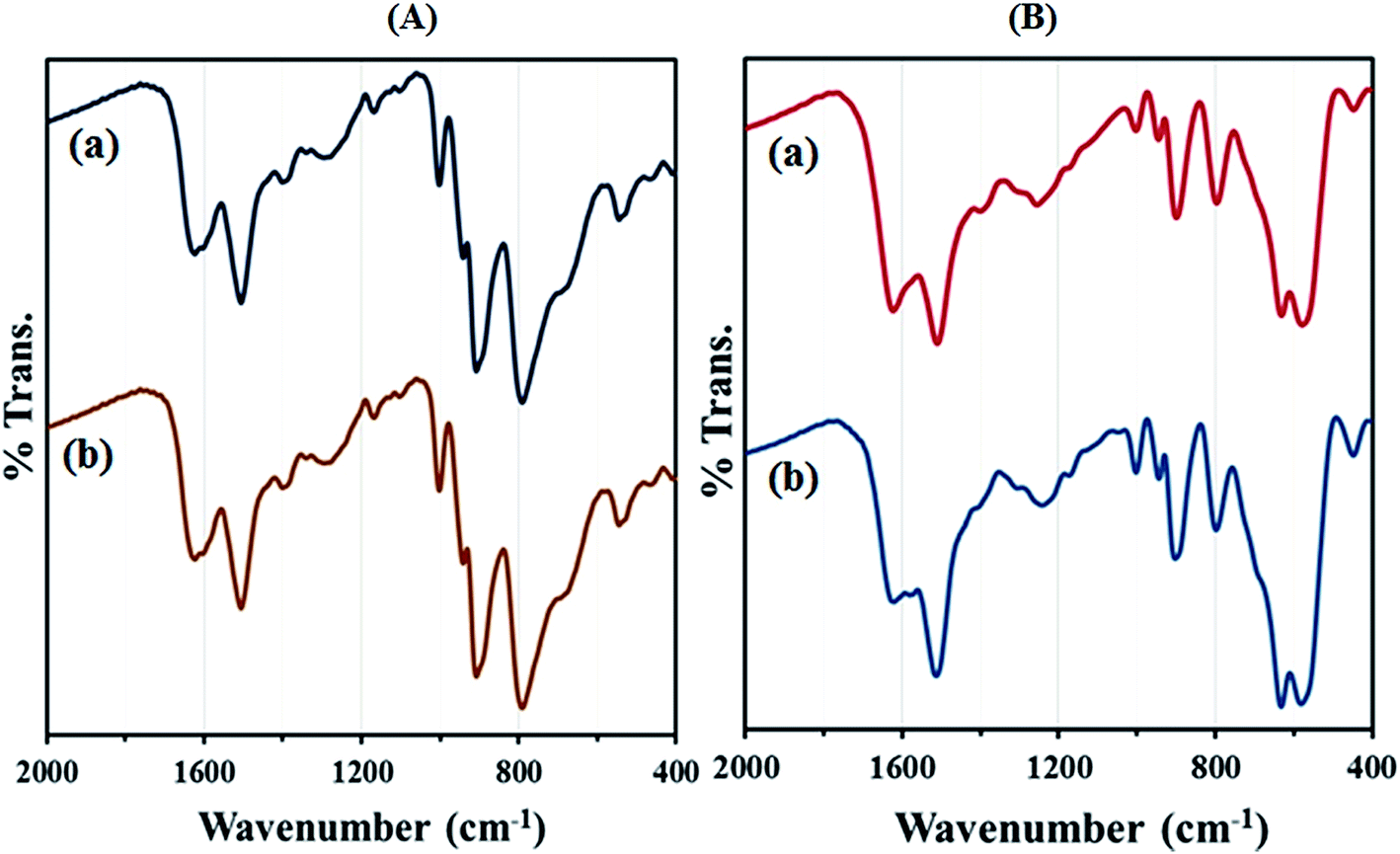 | ||
| Fig. 11 FT-IR spectra of (A) (a) recycled HybPOM for 5 times (b) HybPOM and (B) (a) recycled GO@Fe3O4@HybPOM for 5 times (b) GO@Fe3O4@HybPOM. | ||
The reaction monitoring showed that no further progress of the reaction is observed, confirming that the metal does not leach from the nanocatalyst under hot conditions. The metal Fe, W and Cu leaching from GO@Fe3O4@HybPOM were tested by ICP-MS. The results showed that Fe, W and Cu contents of the GO@Fe3O4@HybPOM after consecutive runs compared with those of the initial GO@Fe3O4@HybPOM were only decreased by less than 1%.
Conclusion
In summary, in this paper we have described (1) a simple and high yield synthesis of new functionalized organic–inorganic polyoxometalates hybrids of H6Cu2[PPD]6[SiW9Cu3O37]·12H2O (HybPOM) and (2) preparation of new GO@Fe3O4@HybPOM nanocomposite by successfully supporting of HybPOM onto graphene oxide decorated with Fe3O4 nanoparticles. HybPOM-functionalized graphene hybrid materials were synthesized by covalently bonding HybPOM onto grapheme oxide via the amide formation and the chemical reaction between amine-functionalized POM and oxygen-containing groups (e.g., epoxy and carboxyl groups) in graphene oxide. The new organic–inorganic polyoxometalates hybrid and nanocomposite were characterized by the elemental analyses, TGA, FT-IR, XPS, XRD, SEM, EDX and AGFM. The results indicate that the size ranges of the nanocomposites are between 20 and 60 nm. The catalytic activity of new NPs was investigated in the Knoevenagel condensation and synthesis of 2,3-dihydroquinazolin-4(1H)-ones. The nano organic–inorganic hybrids and their magnetically reusable nanocomposites were found as effective and easily recoverable heterogeneous catalyst for a wide range of arylaldehydes. Therefore, the newly developed hybrid materials are promising for a wide range of multifunctional applications.Acknowledgements
The authors would like to thank the University of Kurdistan Research Council for their support of this work.References
- (a) M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer, Berlin, 1983, Search PubMed; (b) F. Hussain, U. Kortz, B. Keita, L. Nadjo and M. T. Pope, Inorg. Chem., 2006, 45, 761 CrossRef CAS PubMed.
- Y. Ono, in Perspectives in Catalysis, ed. J. M. Thomas and K. I. Zamaraev, Blackwell, London, 1992, p. 341 Search PubMed.
- (a) I. V. Kozhevnikov, Russ. Chem. Rev., 1987, 56, 811 CrossRef; (b) F. Lefebvre, F. X. Liu-Cai and A. Auroux, J. Mater. Chem., 1994, 4, 125 RSC.
- Y. Izumi, K. Urabe and M. Onaka, Zeolite, Clay and Heteropoly Acids in Organic Chemistry, VCH, Weinheim, Kodansha, Tokyo, 1992 Search PubMed.
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171 CrossRef CAS PubMed.
- N. Mizuno and M. Misono, Chem. Rev., 1998, 98, 199 CrossRef CAS PubMed.
- M. Kooti and M. Afshari, Mater. Res. Bull., 2012, 47, 3473 CrossRef CAS.
- X. M. Yan, J. H. Lei, D. Liu, Y. C. Wu and W. Liu, Mater. Res. Bull., 2007, 42, 1905 CrossRef CAS.
- N. Dubey, S. S. Rayalu, N. K. Labhsetwar and S. Devotta, Int. J. Hydrogen Energy, 2008, 33, 5958 CrossRef CAS.
- K. Zhu, D. Wang and J. Liu, Nano Res., 2009, 2, 1 CrossRef CAS.
- A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009 CrossRef CAS PubMed.
- C. Sanchez and F. Ribot, New J. Chem., 1994, 18, 1007 CAS.
- P. Judeinstein and C. Sanchez, J. Mater. Chem., 1996, 6, 511 RSC.
- S. Liu and Z. Tang, Nano Today, 2010, 5, 267 CrossRef CAS.
- D. Gatteschi, L. Pardi, A. L. Barra and A. Müller, Nature, 1991, 354, 463 CrossRef CAS.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. V. Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064 CrossRef CAS PubMed.
- A. Hu, G. T. Yee and W. Lin, J. Am. Chem. Soc., 2005, 127, 12486 CrossRef CAS PubMed.
- K. Nakada, M. Fujita and G. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 17954 CrossRef CAS.
- H. He, J. Klinowski and M. Forster, Chem. Phys. Lett., 1998, 287, 53 CrossRef CAS.
- J. P. Tessonnier, S. Goubert-Renaudin, Sh. Alia, Y. Yan and M. A. Barteau, Langmuir, 2013, 29, 393 CrossRef CAS PubMed.
- A. H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222 CrossRef CAS PubMed.
- (a) G. M. Scheuermann, L. Rumi, P. Steurer, W. Bannwarth and R. Mulhaupt, J. Am. Chem. Soc., 2009, 131, 8262 CrossRef CAS PubMed; (b) N. R. Shiju and V. V. Guliants, Appl. Catal., A, 2009, 356, 1 CrossRef CAS; (c) L. D. Pachon and G. Rothenberg, Appl. Organomet. Chem., 2008, 22, 288 CrossRef.
- (a) V. Polshettiwar and R. S. Varma, Tetrahedron, 2010, 66, 1091 CrossRef CAS; (b) K. Wang, L. X. Yu, S. Yin, H. Li and H. Li, Pure Appl. Chem., 2009, 81, 2327 CAS; (c) W. Liu, B. J. Li, C. L. Gao and Z. Xu, Chem. Lett., 2009, 38, 1110 CrossRef CAS.
- G. Herve and A. Teze, Inorg. Chem., 1977, 16, 2115 CrossRef CAS.
- M. Hirata, T. Gotou, S. Horiuchi, M. Fujiwara and M. Ohba, Carbon, 2004, 42, 2929 CAS.
- J. L. Xu, M. Li and E. Wang, Mater. Lett., 2002, 54, 303 CrossRef.
- J. Li, X. Zeng, T. Renand and E. V. D. Heide, Lubricants, 2014, 2, 137 CrossRef.
- E. Rafiee, M. Joshaghani, S. Eavani and S. Rashidzadeh, Green Chem., 2008, 10, 982 RSC.
- C. Rocchiccioli-Deltcheff, M. Fournier, R. Franck and R. Thouvenot, Inorg. Chem., 1983, 22, 207 CrossRef CAS.
- K. Singh, A. Ohlan, V. Hung Pham, R. Balasubramaniyan, S. Varshney, J. Jang, S. H. Hur, W. M. Choi, M. Kumar, S. K. Dhawan, B. S. Kongd and J. S. Chung, Nanoscale, 2013, 5, 2411 RSC.
- B. Shen, W. Zhai, M. Tao, J. Ling and W. Zheng, ACS Appl. Mater. Interfaces, 2013, 5, 11383 CAS.
- J. C. Meyer, A. K. Geim, M. I. Katsnelson, K. S. Nosvoselov, T. J. Booth and S. Roth, Nature, 2007, 446, 60 CrossRef CAS PubMed.
- X. Yang, X. Zhang, Y. Ma, Y. Huang, Y. Wang and Y. Chen, J. Mater. Chem., 2009, 19, 2710 RSC.
- L. Salvati, L. E. Makovsky, J. M. Stencel, F. R. Brown and D. M. Hercules, J. Phys. Chem., 1981, 85, 3700 CrossRef CAS.
- S. H. Xuan, W. Q. Jiang, X. L. Gong, Y. Hu and Z. Y. Chen, J. Phys. Chem. C, 2009, 113, 553 CAS.
- Z. H. Ai, L. Z. Zhang, S. C. Lee and W. K. Ho, J. Phys. Chem. C, 2009, 113, 20896 CAS.
- Q. Hua, D. L. Shang, W. H. Zhang, K. Chen, S. J. Chang, Y. S. Ma, Z. Q. Jiang, J. L. Yang and W. X. Huang, Langmuir, 2011, 27, 665 CrossRef CAS PubMed.
- J. H. Zhong, G. R. Li, Z. L. Wang, Y. N. Ou and Y. X. Tong, Inorg. Chem., 2011, 50, 757 CrossRef CAS PubMed.
- H. Zhu, M. Du, D. Yu, Y. Wang, L. Wang, M. Zou, M. Zhang and Y. Q. Fu, J. Mater. Chem. A, 2013, 1, 919 CAS.
- L. F. Tietze, Chem. Rev., 1996, 96, 11 CrossRef.
- T. Sugino and K. Tanaka, Chem. Lett., 2001, 30, 110 CrossRef.
- D. S. Bose and A. V. Narsaiah, J. Chem. Res., Synop., 2001, 3, 36 CrossRef.
- D. J. Connolly, D. Cusack, T. P. O'Sullivan and P. J. Guiry, Tetrahedron, 2005, 61, 10153 CrossRef CAS; N. Isambert, M. M. S. Duque, J. C. Plaquevent, Y. Génisson, J. Rodriguez and T. Constantieux, Chem. Soc. Rev., 2011, 40, 1347 RSC; M. A. P. Martins, C. P. Frizzo, A. Z. Tier, D. N. Moreira, N. Zanatta and H. G. Bonacorso, Chem. Rev., 2014, 114(20), PR1–PR70 CrossRef PubMed; P. Murthy, D. Rambabu, G. R. Krishna, C. M. Reddy, K. R. S. Prasad, M. V. Basaveswara Rao and M. Pal, Tetrahedron Lett., 2012, 53, 863 CrossRef.
- B. V. Subba Reddy, A. Venkateswarlu, C. Madan and A. Vinu, Tetrahedron Lett., 2011, 52, 1891 CrossRef CAS.
- M. Bakavoli, O. Sabzevari and M. Rahimizadeh, Chin. Chem. Lett., 2007, 18, 1466 CrossRef CAS.
- J. M. Khurana and G. Kukreja, J. Heterocycl. Chem., 2003, 40, 677 CrossRef CAS.
- V. B. Rao and C. V. Ratnam, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1979, 18, 409 Search PubMed.
- C. L. Yoo, J. C. Fettinger and M. J. Kurth, J. Org. Chem., 2005, 70, 6941 CrossRef CAS PubMed.
- M. Wang, T. Zhang, Y. Liang and J. Gao, Chin. Chem. Lett., 2011, 22, 1423 CrossRef CAS.
- R. S. Hamid, R. O. Ali and H. Moones, Synth. Commun., 2010, 40, 1231 CrossRef.
- S. Peyman, D. Minoo, A. Z. Mohammad and B. Mostafa, Synlett, 2005, 7, 1155 Search PubMed.
- A. Rostami, B. Tahmasbi, H. Gholami and H. Taymorian, Chin. Chem. Lett., 2013, 24, 211 CrossRef CAS.
- J. X. Chen, W. K. Su, H. Y. Wu, M. C. Liu and C. Jin, Green Chem., 2007, 9, 972 RSC.
- M. Rueping, A. P. Antonchick, E. Sugiono and K. Grenader, Angew. Chem., Int. Ed., 2009, 48, 908 CrossRef CAS PubMed.
- J. A. Cabello, J. M. Campelo, A. Garica, D. Luna and J. M. Marinas, J. Org. Chem., 1984, 49, 5195 CrossRef CAS.
- Y. Q. Cao, Z. Dai, R. Zhang and B. H. Chen, Synth. Commun., 2004, 34, 2965 CrossRef CAS.
- C. Yue, A. Mao, Y. Wei and M. Lu, Catal. Commun., 2008, 9, 1571 CrossRef CAS.
- The Aldrich Library of Infrared Spectra, Aldrich Chemical Co, 3rd edn, 1981 Search PubMed.
- A. K. Mitra, A. De and N. Karchaudhuri, Synth. Commun., 1999, 28, 2731 CrossRef.
- B. M. Choudrary, M. Lakshmi-Kantam, B. Kavita, C. V. Reddy and F. Figueras, Tetrahedron, 2000, 56, 9357 CrossRef.
- A. Kumar, M. Dewan, A. Saxena, A. De and S. Mozumdar, Catal. Commun., 2010, 11, 679 CrossRef CAS.
- B. L. Vilas, P. V. Shinde and M. S. Shingare, Tetrahedron Lett., 2013, 54, 5778 CrossRef.
- S. Rostamizadeh, A. M. Amani, R. Aryan, H. R. Ghaieni and N. Shadjou, Synth. Commun., 2008, 3, 3567 CrossRef.
- A. Ghorbani-Choghamarani and M. Norouzi, J. Mol. Catal. A: Chem., 2014, 395, 172 CrossRef CAS.
- S. S. Parmar, R. Kumar and R. C. Arora, Indian J. Med. Res., 1969, 57, 245 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra15339a |
| This journal is © The Royal Society of Chemistry 2016 |