
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure–composition relationships of bioactive borophosphosilicate glasses probed by multinuclear 11B, 29Si, and 31P solid state NMR
Yang Yu and
Mattias Edén*
Physical Chemistry Division, Department of Materials and Environmental Chemistry, Stockholm University, SE-106 91 Stockholm, Sweden. E-mail: mattias.eden@mmk.su.se
First published on 14th October 2016
Abstract
By combining 11B, 29Si, and 31P nuclear magnetic resonance (NMR) experimental results, we present a comprehensive structural investigation of 15 borophosphosilicate (BPS) glasses of the Na2O–CaO–B2O3–SiO2–P2O5 system: in two base compositions comprising 46 mol% (“S46”) and 49 mol% (“S49”) SiO2, progressive replacements of SiO2 by B2O3 were performed at a constant total Na2O and CaO content. The S46 glass members constitute B-bearing analogs of “45S5 Bioglass” that is utilized extensively for bone grafting in periodontal and orthopedic surgery. Orthophosphate ions prevail throughout all structures, while the silicate network polymerization increases slightly with a growing amount of B2O3 in the glass. 11B NMR revealed continuous BO3 → BO4 conversions for increasing B2O3 content, with asymptotic fractions of 34% and 43% of B[4] coordinations out of the borate speciation observed for the series of S46 and S49 glasses, respectively. While all BPS glasses are homogeneous across a μm-scale, strong preferences for B[3]–O–B[3] and B[4]–O–Si[4] bond formation lead to structures comprising (sub)nm-sized domains of BO3 groups in boroxol rings and borosilicate networks built by SiO4 and BO4 tetrahedra. These borate/borosilicate networks are merged mainly by B[4](3Si) and B[3](1Si) moieties in Si-rich BPS glasses (where each value in parentheses specifies the number of bonds to Si atoms), while B[4](3Si) and B[4](2Si) groups are the dominant network contact points in the B-rich glasses. We discuss the partitioning of non-bridging oxygen ions between the BO3 and SiO4 groups, the relative propensities for B[4]–O–Si[4] and B[4]–O–B[3] bond formation, as well as the expected bearings of our proposed BPS structural model for the glass degradation in aqueous media, where we identify the fractional population of B[4] coordinations and the silicate network connectivity to constitute the dissolution-controlling parameters.
1 Introduction
The elements Al, B, P, and Si build the networks of a vast majority of all oxide-based glasses.1–3 Most of them involve one or two network-forming species, while the development and structural characterization of glasses incorporating three (or more) network formers are much less explored. Yet, they are of interest for the fundamental understanding of glass chemistry, where an improved insight into such mixed-former glass networks will assist the tailoring of their physical, chemical, and mechanical glass properties.Our study concerns borophosphosilicate (BPS) glasses that show great promise for bone-grafting applications.4–14 On their contact with body fluids, such bioactive glasses (BGs)9,15–17 integrate with the bone/tooth by forming a surface layer of hydroxy-carbonate apatite (HCA), whose composition closely mimics bone mineral. Some of the alkali/alkaline-earth-rich borate and borosilicate glasses involved B for Si substitutions of already existing phosphosilicate BGs,4–6,11–13 such as the “19-93”18 and the “45S5 Bioglass®”15,17 compositions; the latter is soda-lime-silica based with molar equivalents 24.6Na2O–26.7CaO–46.1SiO2–2.6P2O5 and has been in clinical use for decades.17 Phosphosilicate BGs feature rings and chains of interconnected silicate tetrahedra (SiO4).3,17 The incorporation of BO3/BO4 borate groups modifies the glass network by the formation of Si–O–B and B–O–B bonds. Compared with phosphosilicate BGs, the borate-based counterparts degrade faster in aqueous media19,20 and are claimed to convert more completely into HCA.4–9,14 The glass degradation is tunable by varying the nB/nSi molar ratio.4,5 Moreover, the dissolution products of B-bearing BGs may promote bone growth and angiogenesis,9,13 as well as stimulating RNA synthesis in fibroblast cells21 to further aid the bone regeneration.
The present report targets two series of Na2O–CaO–B2O3–SiO2–P2O5 glasses (one based on the “45S5” composition15), where SiO2 is replaced by B2O3 at a fixed total Na2O and CaO content. These network modifier-rich (boro)phosphosilicate glasses exhibit fragmented glass networks, mainly built by ring/chain motifs of interlinked SiO4, BO4, and BO3 polyhedra. The local glass structures were probed by a combination of magic-angle spinning (MAS) 11B, 29Si, and 31P NMR experiments. Besides providing the first comprehensive structural account on B-substituted BGs (such as the 45S5 based glasses4,6,13), we demonstrate that P-richer bioactive BPS glasses are feasible to prepare: increasing the P content of a silicate-based BG is believed to promote its HCA formation in vitro17,22,23 and P-rich BPS glass analogs should consequently also be beneficial.
This work also complements the few existing structural reports on BPS glasses that generally targeted the compositional space of low modifier contents relative to P and Si,24–26 encompassing M2O–B2O3–SiO2–P2O5 glasses of low P and alkali metal M = {Na, K} contents24,26 or P-rich compositions of the Na2O–(CaO)–B2O3–SiO2–P2O5 system.25,27 For the latter, P assumes its usual role as a network former in the guise of phosphate chains that are cross-linked to the silicate/borate networks.24,25 This structural scenario is also shared by P-rich M(2)O–SiO2–P2O5 glasses,28,29 but contrasts markedly with that for the fragmented networks of bioactive phosphosilicate glasses, where P is typically a minor component (≲6 mol% P2O5) and predominantly forms orthophosphate (PO43−) anions22,23,30–34 that are charge-balanced by Na+/Ca2+ and distributed randomly at interstitial positions around the silicate network.32,35 This property ensures a rapid release of phosphate moieties when the BG is subjected to (simulated) body fluids.23,33,36 One of the goals of the present work is the verification that this favorable structural role of P remains when substantial amounts of borate groups are incorporated into the glass structure. Previous work on Na2O–B2O3–SiO2–P2O5 glasses26 suggests this not to apply for compositions featuring (significantly) lower Na contents than the total modifier (Na+, Ca2+) reservoirs of the present glasses.
2 Structural features of borophosphosilicate glasses
2.1 Basic building blocks
The present soda-lime BPS glasses involve three traditional glass network-forming species: B, P, and Si. Regarding the structural role of P, earlier reports from Na–B–Si–P–O glasses of relatively low amounts of Na but with comparable Si and P contents as our current glasses revealed a dominance of P2O74− diphosphate groups and P–O–B structural fragments,24,26 suggesting a traditional network-forming role of P. However, as demonstrated in Section 4.1, very few phosphate tetrahedra in the present modifier-rich BPS structures bind to network-forming groups and the P speciation is dominated by orthophosphate anions, denoted Q0P. Henceforth, QnP (QnSi) labels a PO4 (SiO4) tetrahedron with n bridging oxygen (BO) species1–3 that share corners with neighboring tetrahedra. The remaining 4 − n tetrahedral corners represent non-bridging oxygen (NBO) ions that are charge-balanced by electropositive metal ions (herein, Na+ and Ca2+).In contrast with the structure of P-rich (boro)phosphosilicate glasses that also feature SiO5/SiO6 polyhedra,25,28,29 Si is exclusively present in tetrahedral coordination (Si[4]) in modifier-rich/P-poor BPS glasses (see Section 4.2),24–26 where it assumes a set of interconnected {QnSi} groups. Si may also bond to the two B coordinations, B[3] and B[4], which represent trigonal BO3 and tetrahedral [BO4]− moieties, respectively.3,37–39 Here B[p](mSi) labels a B[p] species with m B–O–Si bonds.
The Na+/Ca2+ cations play a dual structural role, where they partially balance the negatively charged [BO4]− and {Q0P, Q1P} moieties, whereas the remaining cations are termed network modifiers because they reduce the glass network polymerization by breaking Si/B[3]–O–Si/B[3] bonds, thereby driving BO → NBO conversions.1–3 The polymerization degree associated with a network-forming species E may be described by its average number of BO atoms per polyhedron (![[N with combining macron]](https://www.rsc.org/images/entities/i_char_004e_0304.gif) EBO),3,23,33,40 which is often referred to as the network connectivity.22,41,42 For silicate-based glasses, the silicate network connectivity (SiBO) may be derived from the set {xnSi} accessible by 29Si NMR.1–3 Herein, the symbol “x” is reserved for fractions, where xnE labels the fractional population of QnE out of the entire {QnE} speciation (0 ≤ n ≤ 4).
EBO),3,23,33,40 which is often referred to as the network connectivity.22,41,42 For silicate-based glasses, the silicate network connectivity (SiBO) may be derived from the set {xnSi} accessible by 29Si NMR.1–3 Herein, the symbol “x” is reserved for fractions, where xnE labels the fractional population of QnE out of the entire {QnE} speciation (0 ≤ n ≤ 4).
Concerning the B partitioning among B[3]/B[4] coordinations and their precise structural relationship relative to the silicate tetrahedra, no clear consensus is reached for BPS glasses24–27 and the compositional space relevant for bioactive glasses remains essentially unexplored. Yet, these aspects have been intensively investigated for the limiting borosilicate systems,2,3,6,37,39,43–54 notably so for Na2O–B2O3–SiO2 glasses. For the latter stoichiometric compositions parametrized as RNa2O–B2O3–KSiO2, Bray and co-workers37,38 proposed a structural model that provides the relative {B[4], B[3]} populations for variable molar ratios K and R; it is henceforth referred to as the Yun–Dell–Bray–Xiao (YDBX) model. It predicts that for low Na2O contents (i.e., small R-values), B[3] → B[4] conversions occur up to R ≤ Rmax = K/16 + 1/2,38 meaning that Na+ solely acts to charge-balance the [BO4]− tetrahedra. Consequently, for all compositions with R < Rmax, the borosilicate structure only comprises B[3]–O–B[3], Si–O–Si, and B[4]–O–Si bonds, the latter forming B[4](OSi)4 groups. NBO species only exist when R > Rmax, where they initially enter solely at the SiO4 tetrahedra, whereas the BO3 groups starts accommodating NBO ions for Na-richer compositions with R > (K + 1)/4.38
The YDBX structural picture provides reasonable predictions of the B speciation in Na–B–Si–O glasses, and sometimes also for some other M+/M2+ cation-bearing amorphous borosilicates. However, the model was formulated solely with insight from 11B NMR, and subsequent studies indicate its limitations: overall, the borosilicate glass structure is less ordered than suggested by the YDBX model and the NBO ions are more evenly distributed among SiO4 and BO3 groups45,46,48–53 compared with the YDBX scenario. Moreover, the {BO3, BO4, SiO4} network building blocks are markedly more intermixed, with all types of B[3]/B[4]–O–B[3]/Si linkages encountered,39,46–54 albeit the preferences of B–O–Si bond formation decreases in the order B[4] > B[3](non-ring) > B[3](ring).50–52 Here “ring” specifies BO3 groups that exclusively form boroxol (B3O6) rings, as present in vitreous B2O3 and borate glasses,2,3,39,55 whereas “non-ring” implies linkages to BO4/SiO4 tetrahedra.50–54,56 To avoid local negative charge accumulation, [BO4]− tetrahedra involve solely BO (as opposed to NBO) species,3,37,38,40,53 while B[4]–O–B[4] bridges are strongly disfavored, or even absent.1–3,37,38,50–53 Herein, we explore the composition-dependence of the B speciation in the more complex soda-lime BPS glass system, targeting NBO-richer networks than those thoroughly investigated in the ternary/quaternary M(2)O–(M′O)–B2O3–SiO2 systems.
2.2 Our borophosphosilicate glass design
Table 1 lists our targeted Na2O–CaO–B2O3–SiO2–P2O5 glass compositions. Each is labeled SNp(q), where N is the sum of SiO2 and B2O3 contents in mol%, while p and q represent the mol% of P2O5 and B2O3, respectively. Note that S462.6(0) corresponds to the widely utilized “45S5 Bioglass” composition.15,17 When collectively referring to all its members, we employ the nomenclature “S46” and “S49” for the two series, which mainly differ in (i) the Si contents of the two base compositions, S462.6(0) and S494.0(0), and thereby their glass-network polymerization degrees reflected in the corresponding silicate network connectivities22,23,33SiBO = 2.11 and SiBO = 2.54; (ii) the P2O5 contents that are constant at 2.6 mol% and 4.0 mol% within each respective set; (iii) the relative Na/Ca amounts, with the molar ratios nNa/nCa = xNa/xCa being 1.84 and 2.08, respectively, while the sum x(Na2O) + x(CaO) remains constant within each S46 and S49 family, where xE or x(E) denotes the molar fraction of species E.
| Label | fb/% | x(Na2O) | x(CaO) | x(SiO2) | x(B2O3) | x(P2O5) | Stoichiometric formulac | xNa/xCad | xB/xSid | R′ e | K f |
|---|---|---|---|---|---|---|---|---|---|---|---|
a Each glass is labeled SNp(q), where N = 100[x(SiO2) + x(B2O3)] is the sum of mol% of SiO2 and B2O3, whereas p and q are the mol% of P2O5 and B2O3, respectively.b f = 100x(B2O3)/[x(SiO2) + x(B2O3)] is the percentage of B2O3 substitution for SiO2.c Stoichiometric formula with coefficients specified as atom fractions that sum to unity.d Ratios of the atomic fractions of Na and Ca (xNa/xCa), and B and Si (xB/xSi), respectively.e Ratio 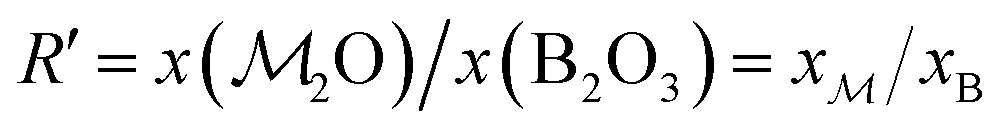 , with , with 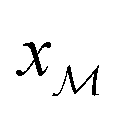 calculated from eqn (1).f K = x(SiO2)/x(B2O3). calculated from eqn (1).f K = x(SiO2)/x(B2O3). |
|||||||||||
| S462.6(0) | 0 | 0.246 | 0.267 | 0.461 | 0.000 | 0.026 | Na0.173Ca0.094Si0.162B0.000P0.018O0.552 | 1.84 | 0.00 | — | — |
| S462.6(5) | 10 | 0.246 | 0.267 | 0.415 | 0.046 | 0.026 | Na0.168Ca0.091Si0.142B0.031P0.018O0.550 | 1.84 | 0.22 | 9.46 | 9.00 |
| S462.6(9) | 20 | 0.246 | 0.267 | 0.369 | 0.092 | 0.026 | Na0.163Ca0.088Si0.122B0.061P0.017O0.548 | 1.84 | 0.50 | 4.73 | 4.00 |
| S462.6(14) | 30 | 0.246 | 0.267 | 0.322 | 0.138 | 0.026 | Na0.158Ca0.086Si0.104B0.089P0.017O0.547 | 1.84 | 0.86 | 3.15 | 2.33 |
| S462.6(18) | 40 | 0.246 | 0.267 | 0.277 | 0.184 | 0.026 | Na0.153Ca0.083Si0.086B0.115P0.016O0.546 | 1.84 | 1.33 | 2.37 | 1.50 |
| S462.6(28) | 60 | 0.246 | 0.267 | 0.184 | 0.277 | 0.026 | Na0.145Ca0.079Si0.054B0.163P0.015O0.543 | 1.84 | 3.00 | 1.58 | 0.67 |
| S462.6(37) | 80 | 0.246 | 0.267 | 0.092 | 0.369 | 0.026 | Na0.138Ca0.075Si0.026B0.206P0.015O0.541 | 1.84 | 8.00 | 1.18 | 0.25 |
| S494.0(0) | 0 | 0.241 | 0.233 | 0.486 | 0.000 | 0.040 | Na0.165Ca0.079Si0.166B0.000P0.027O0.562 | 2.08 | 0.00 | — | — |
| S494.0(2) | 5 | 0.241 | 0.233 | 0.462 | 0.024 | 0.040 | Na0.162Ca0.078Si0.155B0.016P0.027O0.561 | 2.08 | 0.11 | 14.7 | 19.0 |
| S494.0(5) | 10 | 0.241 | 0.233 | 0.437 | 0.049 | 0.040 | Na0.160Ca0.077Si0.145B0.032P0.026O0.560 | 2.08 | 0.22 | 7.37 | 9.00 |
| S494.0(7) | 15 | 0.241 | 0.233 | 0.413 | 0.073 | 0.040 | Na0.157Ca0.076Si0.134B0.047P0.026O0.559 | 2.08 | 0.35 | 4.91 | 5.67 |
| S494.0(10) | 20 | 0.241 | 0.233 | 0.389 | 0.097 | 0.040 | Na0.155Ca0.075Si0.125B0.062P0.026O0.558 | 2.08 | 0.50 | 3.69 | 4.00 |
| S494.0(15) | 30 | 0.241 | 0.233 | 0.340 | 0.146 | 0.040 | Na0.150Ca0.072Si0.106B0.091P0.025O0.557 | 2.08 | 0.86 | 2.46 | 2.33 |
| S494.0(19) | 40 | 0.241 | 0.233 | 0.292 | 0.194 | 0.040 | Na0.146Ca0.070Si0.088B0.117P0.024O0.555 | 2.08 | 1.33 | 1.85 | 1.50 |
| S494.0(24) | 50 | 0.241 | 0.233 | 0.243 | 0.243 | 0.040 | Na0.141Ca0.068Si0.071B0.142P0.023O0.553 | 2.08 | 2.00 | 1.48 | 1.00 |
The precise glass forming region of the complex Na2O–CaO–B2O3–SiO2–P2O5 system is unknown and is currently being explored by us. Preliminary results indicate that the S49 series may allow higher B2O3 substitution levels than the S494.0(24) glass included in the present NMR study. However, it remains unclear how much further the amount of P may be increased for BPS glasses with significant B contents.
As demonstrated in Section 4.1, the phosphate speciation in our Na2O–CaO–B2O3–SiO2–P2O5 glasses is dominated by orthophosphate (Q0P) groups (0.86 ≤ x0P ≤ 0.96), with the remaining constituting Q1P moieties. To assist comparisons with the YDBX37,38 prediction of the B[3]/B[4] partitioning, we introduce the ratio 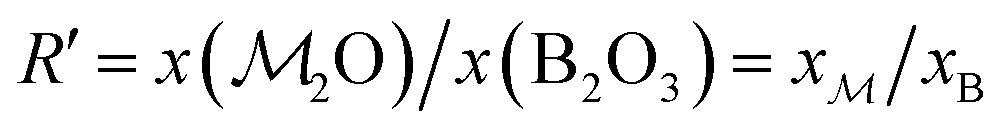 that accounts for the consumption of Na+/Ca2+ ions required to charge-balance all {Q0P, Q1P} phosphate groups by considering the fictive monovalent ion
that accounts for the consumption of Na+/Ca2+ ions required to charge-balance all {Q0P, Q1P} phosphate groups by considering the fictive monovalent ion 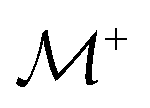 , whose content relates to the net amount of positive charges available for the B/Si species of the borosilicate network, according to
, whose content relates to the net amount of positive charges available for the B/Si species of the borosilicate network, according to
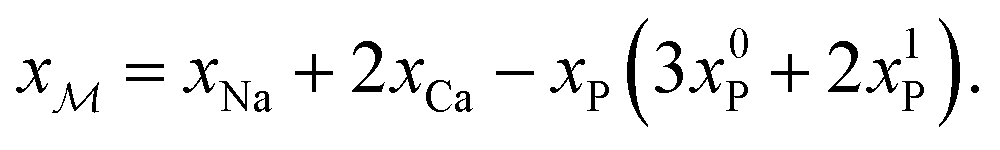 | (1) |
Consequently, each Na2O–CaO–B2O3–SiO2–P2O5 glass composition SNp(q) maps onto a 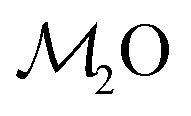 –B2O3–SiO2 counterpart associated with the parameter R′ that may be contrasted directly with the result of a Na2O–B2O3–SiO2 analog with R = x(Na2O)/x(B2O3) and K = x(SiO2)/x(B2O3) from the YDBX model.37,38 Table 1 lists the resulting values of R′, K, and other relevant glass composition factors, such as xB/xSi.
–B2O3–SiO2 counterpart associated with the parameter R′ that may be contrasted directly with the result of a Na2O–B2O3–SiO2 analog with R = x(Na2O)/x(B2O3) and K = x(SiO2)/x(B2O3) from the YDBX model.37,38 Table 1 lists the resulting values of R′, K, and other relevant glass composition factors, such as xB/xSi.
Note that this simplistic picture only accounts for the total positive charge reservoir provided from the monovalent Na+ and divalent Ca2+ cations. In our previous NMR studies of 31P and 29Si environments in phosphosilicate glasses,33–35 no significant differences were observed from slight variations in the relative Na/Ca contents among different glasses, where the main glass-network alterations are captured well by solely considering the total glass modifier content, the P content, and the silicate network connectivity. Also for the present BPS glasses, the minor difference among the xNa/xCa ratios of the S46 and S49 families is not expected to alter the glass structures significantly; indeed, we verified for two S49 members that the two xNa/xCa values of 1.84 and 2.08 yield B[4] fractional populations that agree within 97% [for S494.0(15)] and 99% [for S494.0(24)]. While such minor differences are immaterial for any result discussed herein, further studies are required to assess the precise structural bearings from the relative amounts of Na+ and Ca2+ (if any), as well as their influences on the in vitro glass degradation and bioactivity.
3 Materials and methods
3.1 Glass preparation and characterization
6.0 g batches of the BPS glasses were prepared by a standard melt-quench method, using precursors of SiO2 (99.99%), Na2CO3 (99.99%), and CaCO3 (99.9%) from ChemPur, and NaH2PO4 (99.99%, Merck), and H3BO3 (99.9%, Sigma). Prior to its weighing, the SiO2 powder was heated at 950 °C for 24 h to remove potential OH/H2O contaminations. The precursors were ground, weighted, and transferred to a bottle that was shaken thoroughly before another mixing stage in a mortar for 1 hour to achieve homogeneous powders. The mixture was then placed in a Pt crucible and heated in an electric furnace at 950 °C for 120 min to allow for complete CO2 removal. Depending on the P2O5 and SiO2 contents of each batch, the temperature was then increased to 1200–1250 °C (S46 series) and 1300–1400 °C (S49 series), held for 20 min (S46 specimens) or 30 min (S49), before quenching the melt by immersing the bottom of the crucible in water.The evaporation losses during synthesis remained ≲1.5 wt% throughout. We verified by 11B MAS NMR on two specimens [S462.6(28) and S462.6(37)] that insignificant alterations in the peakshapes and NMR intensities resulted after repeating the melt-quench process on the initially prepared glasses. Besides confirming sufficient precursor mixing, it strongly suggests very limited evaporation losses of the most volatile glass ingredient (i.e., B). Moreover, the relative integrated 31P NMR intensities (Section 4.1) among the samples were in excellent agreement with the corresponding nominal x(P2O5) values, thereby further suggesting intact batch compositions.
Polished glass samples were coated by a 10–20 nm thick carbon layer and examined by scanning electron microcopy (SEM), using a JEOL JSM-7000F microscope in backscatter electron mode at a 15 kV acceleration voltage. None of the specimens manifested signatures of phase separation down to the shortest accessible length-scale of 1 μm.
3.2 Solid-state NMR
All NMR experiments utilized Bruker Avance-III spectrometers. The 31P and 29Si MAS NMR spectra were recorded at a magnetic field (B0) of 9.4 T, which corresponds to 31P and 29Si Larmor frequencies of −162.0 MHz and 79.5 MHz, respectively. Glass powder were filled in 7 mm zirconia rotors that were spun at 7.00 kHz during the NMR signal acquisitions. All 11B NMR experimentation was performed at B0 = 14.1 T (−192.5 MHz 11B Larmor frequency) with 3.2 mm rotors spinning at 24.00 kHz.The 31P and 29Si NMR acquisitions were performed in blocks, using single-pulse excitation with flip angles of 70° (31P) or 60° (29Si) at radio-frequency (rf) nutation frequencies νnut = |γ|B1/2π ≈ 60 kHz, where B1 is the rf amplitude. The 31P NMR acquisitions involved ≈48 accumulated signal transients, with 5000 s “equilibration” delays before the start of each block of 8 transients, and relaxation delays of 2500 s between each rf pulse within the block. Depending on the Si content of the glass, between 92–156 transients were recorded with 3600 s relaxation delays and equilibration delays of 3.5 h between each block of 4–8 transients. 300 Hz and 200 Hz full width at half maximum (fwhm) height Gaussian broadening were employed in the 29Si data processing of the S46 and S49 glasses, respectively, whereas no signal apodization was employed for the 31P NMR data.
11B MAS NMR spectra were recorded using short (0.5 μs; ≈15°) rf pulses at νnut ≈ 75 kHz, relaxation delays of 15 s, and 512–3072 accumulated transients, depending on the B content of the sample. 11B background signals from the MAS NMR probehead were removed by subtracting the results from an empty rotor. No signal apodization was used in the processing, except for the S494.0(2) glass that employed 225 Hz fwhm Gaussian broadening.
Triple quantum MAS (3QMAS)57 11B NMR acquisitions employed the Z-filter scheme58,59 utilizing the symmetric 0 → ±3 → ±1 → 0 → −1 quantum coherence pathways. All rf pulses for 3Q coherence (3QC) excitation and conversion operated at νnut = 115 kHz. The 3QC excitation pulse lasted for 5.8 μs. Two FAM60 blocks with equal durations of the pulses and delays of 0.90 μs were used for the ±3 → ±1 coherence transfer. The two central transition (CT) selective 90° pulses of duration 17.0 μs (νnut = 7.4 kHz) following the 3QC conversion were interleaved by a Z-filter delay of one rotational period. The 2D NMR acquisitions involved the States procedure61 for providing absorptive peaks with frequency sign-discrimination along the indirect spectral dimension, together with dwell times {Δt1, Δt2} = {41.7, 6.1} μs, 2 s relaxation delays, and 288–3456 co-added transients. Typically, 100(t1) × 2428(t2) time-points were recorded and zero-filled to 512 × 8192 points. The indirect 3QMAS dimension was processed according to the “Cz” convention.57,62
These experimental conditions—i.e., the flip angles, “pre-equilibration” and “relaxation” (or “interpulse”) delays—were selected for each nucleus from separate saturation-recovery T1 measurements to ensure NMR data quantitatively reflecting the populations of the various B, Si and P structural sites. 31P, 11B, and 29Si shifts are quoted relative to 85% H3PO4(aq.), neat BF3·OEt2, and neat tetramethylsilane (TMS), respectively; the liquids were also exploited for estimating rf nutation frequencies, except for the 31P rf pulses that were calibrated directly on the glass samples.
4 Results
4.1 31P MAS NMR
The 31P MAS NMR spectra recorded from the S46 and S49 glass families are shown in Fig. 1. The two base glasses, S462.6(0) and S494.0(0), reveal peak maxima at 8.7 ppm and 8.3 ppm, respectively, corresponding to signals from orthophosphate groups charge-balanced by Na+/Ca2+ cations.22,30–34,63 The marginally lower shift of the NMR peak from the S494.0(0) glass, which features a slightly higher xNa/xCa ratio andSiBO value, stems from two counteracting factors: the deshielding effect of Na+ (relative to Ca2+) that increases the shift,22,30,33,34,63 and the shielding (shift reduction) resulting from emphasized Ca2+–PO43− associations when SiBO grows.34 The large fwhm of the NMR peaks ≈7.5 ppm reflects the disordered glass structures. The 31P resonances of S462.6(0) are characterized by a nearly Gaussian peakshape, while that for S494.0(0) appears more asymmetric, featuring a “tail” towards lower shifts: it stems from Q1P moieties, i.e., phosphate groups with one P–O–Si (as opposed to P–O–P) bond.32–34,36,64,65 For both S46 and S49 series, Fig. 1 reveals progressively more asymmetric 31P resonances for increasing x(B2O3) in the glass. While the growing amount of Q1P species may potentially involve either of P–O–Si/B bonding scenarios, additional heteronuclear 31P{11B} NMR experiments (to be published elsewhere) suggest that few P–O–B bonds form and that P–O–Si bridges prevail, as in the parent B-free phosphosilicate glasses.
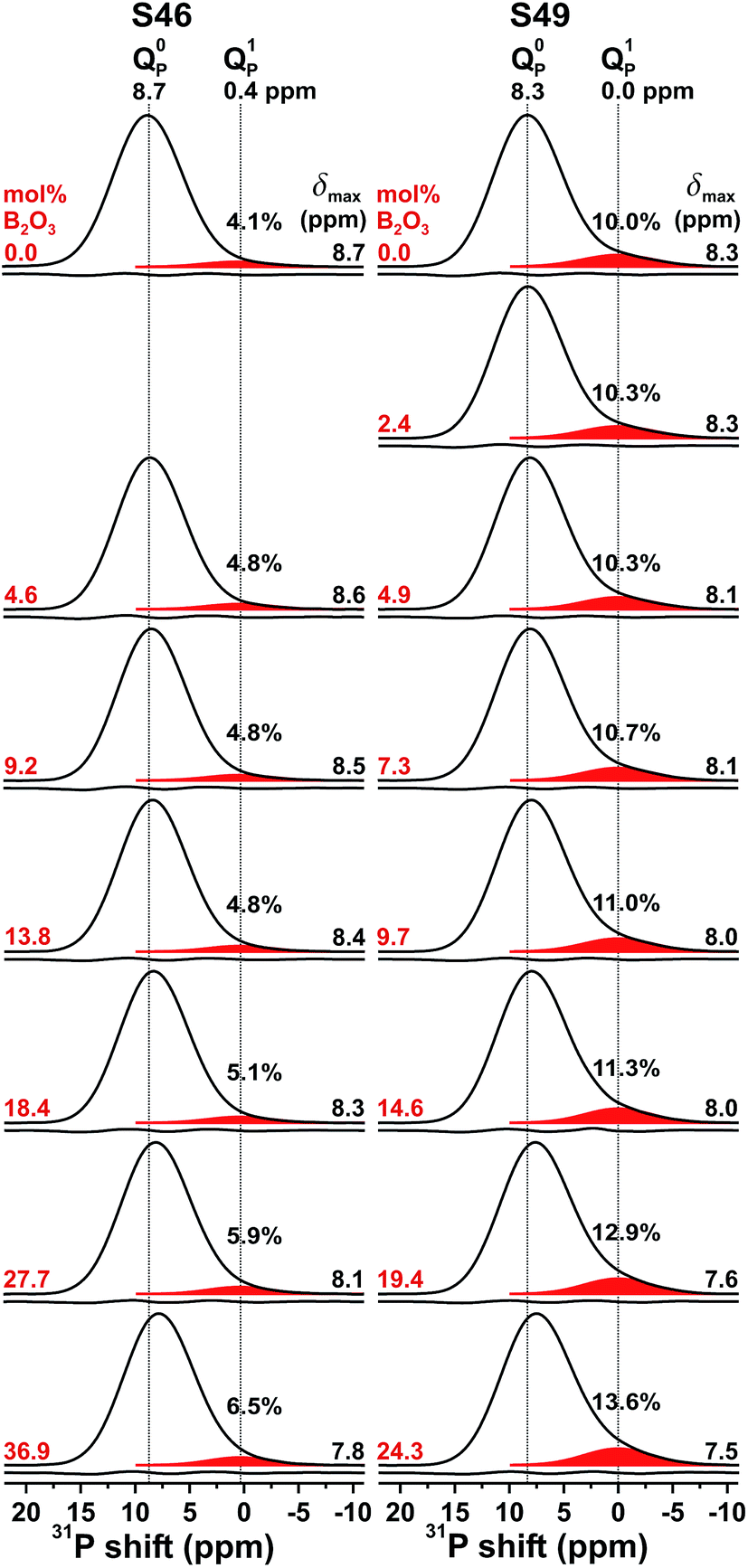 | ||
| Fig. 1 Experimental 31P MAS NMR spectra recorded at 9.4 T from the as-indicated SNp(q) glasses, with members from the S46 and S49 series shown in the left and right panels, respectively, and listed according to increasing B2O3 content from top to bottom. The 31P resonances from the Q0P (main peak) and Q1p (minor) groups are indicated, with the fractional populations of the latter specified in %. The curve beneath each NMR spectrum represents the difference between experimental and best-fit data. | ||
To quantify the influence of the increased B2O3 content on the phosphate speciation, we deconvoluted each 31P NMR peak into two Gaussian components by a constrained iterative fitting, considering both the centerband and first-order spinning sidebands stemming from each Q0P/Q1P group (see Mathew et al.33). Each peak component was represented by three parameters, {δnP, WnP, xnP}, corresponding to the mean 31P chemical shift (δnP), the fwhm (WnP), and the fractional population (xnP). Table 2 lists the best-fit results. For both the S46/S49 glass series, the chemical shift of the orthophosphate groups (δ0P) decreases congruently with the respective maximum of the net NMR signal, suggesting a more pronounced propensity for the Q0P groups to associate with the Ca2+ cations relative to Na+.33,34,36 The more than twice as large x1P population (≈10%) of the S494.0(0) structure relative to its S462.6(0) counterpart is consistent with its larger SiBO value and the linear SiBO/x1P relationship established recently by Mathew et al.33 When the B2O3 content grows, a weak increase of x1P is observed between 0.041–0.065 (for S46) and 0.100–0.136 (for S49), which reflect minor increases of the silicate network polymerization, as discussed in Section 4.2.
| Label | 31P NMR dataa | 29Si NMR datab | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| δPmax (ppm) | WP (ppm) | δ0P (ppm) | W0P (ppm) | x0P | δ1P (ppm) | W1P (ppm) | x1P | δSimax (ppm) | δSiCG (ppm) | WSi (ppm) | |
| a Shift at peak maximum (δPmax; uncertainty ±0.08 ppm) and full width at half maximum (fwhm) height (WP; ±0.15 ppm) of the net 31P NMR signal, as well as the chemical shift (δnP; ±0.3 ppm), fwhm height (WnP; ±0.5 ppm), and fractional population (xnP; ±0.01) of each QnP contribution extracted by NMR spectra deconvolution.b Peak maximum (δSimax; uncertainty ±0.15 ppm), the center-of-gravity shift (δSiCG; ±0.25 ppm), and fwhm height (WSi; ±0.25 ppm) of the net 29Si NMR signal. | |||||||||||
| S462.6(0) | 8.72 | 7.50 | 8.7 | 7.4 | 0.959 | 0.4 | 7.3 | 0.041 | −79.5 | −81.0 | 11.1 |
| S462.6(5) | 8.60 | 7.35 | 8.6 | 7.2 | 0.952 | 0.4 | 7.1 | 0.048 | −80.0 | −80.7 | 11.9 |
| S462.6(9) | 8.46 | 7.37 | 8.5 | 7.2 | 0.952 | 0.4 | 7.1 | 0.048 | |||
| S462.6(14) | 8.35 | 7.41 | 8.4 | 7.3 | 0.952 | 0.4 | 6.8 | 0.048 | −79.9 | −80.5 | 12.4 |
| S462.6(18) | 8.27 | 7.44 | 8.3 | 7.3 | 0.949 | 0.4 | 6.8 | 0.051 | |||
| S462.6(28) | 8.07 | 7.53 | 8.1 | 7.4 | 0.941 | 0.4 | 6.8 | 0.059 | −79.5 | −80.1 | 12.7 |
| S462.6(37) | 7.79 | 7.57 | 7.8 | 7.4 | 0.935 | 0.4 | 6.6 | 0.065 | −78.6 | −79.6 | 12.5 |
| S494.0(0) | 8.34 | 7.46 | 8.3 | 7.3 | 0.900 | 0.0 | 7.8 | 0.100 | −82.5 | −84.9 | 16.0 |
| S494.0(2) | 8.28 | 7.49 | 8.3 | 7.3 | 0.897 | 0.0 | 7.8 | 0.103 | −82.8 | −84.4 | 15.7 |
| S494.0(5) | 8.14 | 7.52 | 8.1 | 7.3 | 0.897 | 0.0 | 7.6 | 0.103 | |||
| S494.0(7) | 8.08 | 7.55 | 8.1 | 7.3 | 0.893 | 0.0 | 7.6 | 0.107 | −83.2 | −84.2 | 15.0 |
| S494.0(10) | 7.96 | 7.59 | 8.0 | 7.4 | 0.890 | 0.0 | 7.5 | 0.110 | |||
| S494.0(15) | 7.97 | 7.66 | 7.9 | 7.4 | 0.887 | 0.0 | 7.2 | 0.113 | |||
| S494.0(19) | 7.64 | 7.76 | 7.6 | 7.5 | 0.871 | 0.0 | 7.3 | 0.129 | |||
| S494.0(24) | 7.51 | 7.85 | 7.5 | 7.5 | 0.864 | 0.0 | 7.3 | 0.136 | −82.9 | −84.0 | 14.3 |
4.2 29Si MAS NMR
The 29Si chemical shift of a QnSi group in a silicate glass depends essentially on the same structural factors as 31P in phosphate glasses (see Section 4.1), i.e., on the relative number of BO/NBO species at the SiO4 tetrahedron (n) and the precise Na+/Ca2+ constellation that provides local charge balance.1–3,40,66,67 However, in a multicomponent glass—such as the present Na2O–CaO–SiO2–P2O5 specimens S462.6(0) and S494.0(0)—the BO/NBO distribution, variations in the relative numbers of Na+/Ca2+ cations around each SiO4 group, and distributions in Si–O bond lengths and Si–O–Si bond angles give a plethora of co-existing local 29Si environments with similar chemical shifts that preclude the identification of the responses from individual QnSi species.1–3,40Fig. 2(a) and (b) presents the 29Si MAS NMR spectra of the two B-free S462.6(0) and S494.0(0) glasses. Consistent with the expected domination of Q2Si groups in the S462.6(0) structure (SiBO = 2.11), its 29Si NMR peakshape is nearly Gaussian. However, a “tail” towards lower shifts is discernible, signifying the presence of Q3Si groups resonating at lower chemical shifts ≈−88 ppm. Their larger contributions to the more polymerized silicate network of the S494.0(0) glass is evident from its NMR peakshape that corresponds to a superposition of (mainly) two Gaussian peaks centered around −80 ppm (Q2Si) and −88 ppm (Q3Si).
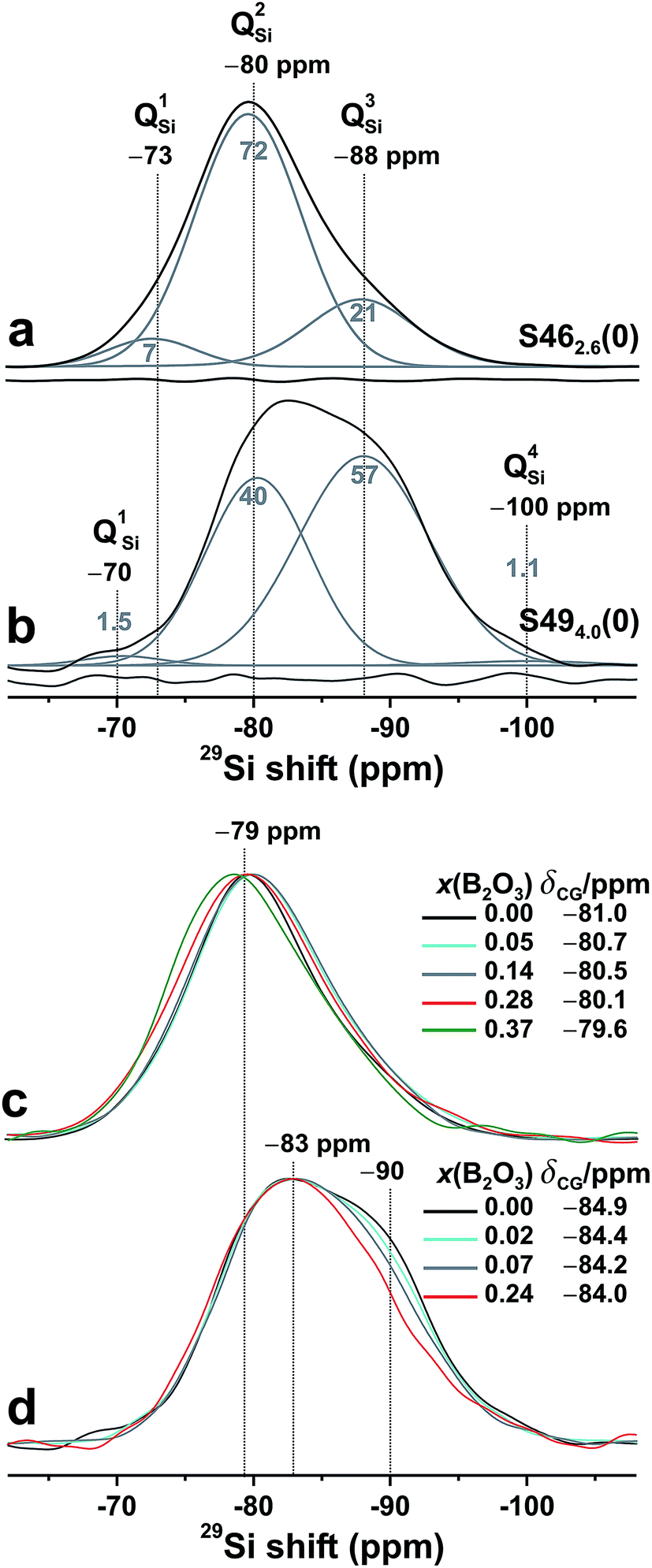 | ||
| Fig. 2 Experimental 29Si MAS NMR spectra obtained at 9.4 T (black traces) from the (a) S462.6(0) and (b) S494.0(0) glasses, shown together with the set of {QnSi} component peaks (gray traces) obtained by spectra deconvolution. Each gray number within (or on top) of each NMR peak specifies its relative population in %. The curve beneath each NMR spectrum represents the difference between the experiment and the corresponding best-fit. (c and d) NMR spectra obtained from glasses with as-specified molar fractions of B2O3 from each (c) S46 and (d) S49 series. Note that the results for S462.6(0)—i.e., the “45S5 Bioglass”15—are reproduced from Mathew et al.33 | ||
Fig. 2(a) and (b) also displays the deconvolution of each NMR spectrum into its underlying {QnSi} peak components, where the results for S462.6(0) are reproduced from Mathew et al.33 Q2Si groups (≈72%) dominate the silicate speciation of the S462.6(0) structure, as expected from its nominal silicate network connectivity of 2.11. Indeed, the best-fit {x1Si, x2Si, x3Si} populations together with the expression
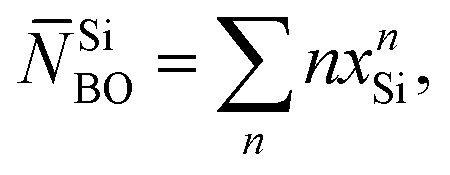 | (2) |
SiBO = 2.14;33 see Fig. 2(a). The respective {x1Si, x2Si, x3Si, x4Si} values of S494.0(0) are {0.015, 0.403, 0.571, 0.011}, corresponding to SiBO = 2.58 and in excellent agreement with the nominal value SiBO = 2.54. These populations accord well with those of the B-free Na–Ca–Si–P–O “BG4.0(2.5)” glass discussed in ref. 33 that is associated with SiBO = 2.50. The present S494.0(0) composition features very close SiO2 and P2O5 contents to BG4.0(2.5), with the main distinction being their different Na/Ca contents, where S494.0(0) is richer in Na (xNa/xCa = 2.08) relative to BG4.0(2.5) with xNa/xCa = 1.54.
Fig. 2(c) and (d) displays the 29Si NMR spectra of the B-bearing members of each S46/S49 glass family. Only minor resonance-alterations are observed when SiO2 is substituted by B2O3. Notably, the NMR peak maxima from all S46 glasses remain ≈−80 ppm for x(B2O3) ≤ 0.28, where only the B-richest glass reveals a noticeable peak displacement to −78.6 ppm. Likewise, all S49-deriving glasses manifest equal peak maxima ≈−83 ppm, with their main distinctions being a reduced signal intensity in the low-ppm region ≈−90 ppm as the B content is increased. Two factors may account for the minor increase of the 29Si chemical shifts when B is included in the glass structure:
(i) The silicate network polymerization decreases slightly, such that lower-n QnSi populations grow, thereby inducing a 29Si deshielding with a typical shift displacement of 7–12 ppm per QnSi → Qn−1Si transformation.1–3,66,67 However, this implies an increase of the average number of NBO species at the SiO4 groups. This scenario is precluded by the constant Na+/Ca2+ content of the glasses of each S46/S49 series: after accounting for the modifier consumption by the phosphate and [BO4]− species, the BO3 groups may also accommodate NBO ions and therefore an increase (rather than a decrease) of the silicate network polymerization is anticipated.
(ii) Substitutions of Si by B[4] species in the second coordination shell of 29Si typically induce ≈5 ppm 29Si deshielding per 29Si–O–Si → 29Si–O–B[4] conversion,1–3,39,45–47 whereas the 29Si–O–Si → 29Si–O–B[3] replacements preserve the net charge of the O atom, thereby leaving the 29Si chemical shift value essentially invariant.43,45–47 While it is then tempting to interpret the minor alterations of the 29Si NMR spectra in Fig. 2 to reflect a predominance of Si–O–B[3] bonds,39 this scenario is excluded from the 11B NMR results discussed in Sections 4.3 and 5.1.
Most likely, the weak but clearly discernible deshielding of the 29Si nuclei reflects the net effect of the two opposing factors (i) and (ii), i.e., the shift elevation from the neighboring B[4] atoms is partially offset by a slight shift reduction from a higher SiBO value. The enhanced Si/B intermixing leads to a larger shift dispersion stemming from a plethora of 29Si environments featuring variable number of Si–O–B[4] bonds;40,45–47 see Fig. 2(d). The 29Si resonance-broadening and overall loss of peakshape features for increasing B2O3 content is well documented: for instance, see Fig. 3 of El-Damrawi et al.46 and Fig. 4 of Edén et al.40 Owing to the multitude of potentially co-existing but unresolved 29Si NMR responses, spectra deconvolutions are unfortunately not warranted for the B-bearing glasses. However, given the absence of significant 31P–O–B contacts (Section 4.1) coupled with the linear dependence of the Q1P population on the average silicate network connectivity,33 we may estimate SiBO from the x1P-values of Table 2 and the quantitative x1P/SiBO relationship established by Mathew et al.33 Across the S46 glass series, the growth of x1P from 0.041 to 0.065 then translates into a minor increase of SiBO from 2.11 to ≈2.24, while the S49 glasses yield the corresponding SiBO growth from 2.54 to 2.73. Hence, when the B2O3 content of the glass elevates, the silicate network polymerization increases slightly, while the 29Si sites become slightly deshielded by the formation of Si–O–B[4] bonds. The observation of a nearly constant silicate network polymerization on B ↔ Si substitutions at a constant modifier content accords with the recent findings by Smedskjaer et al.53 for more condensed borosilicate networks.
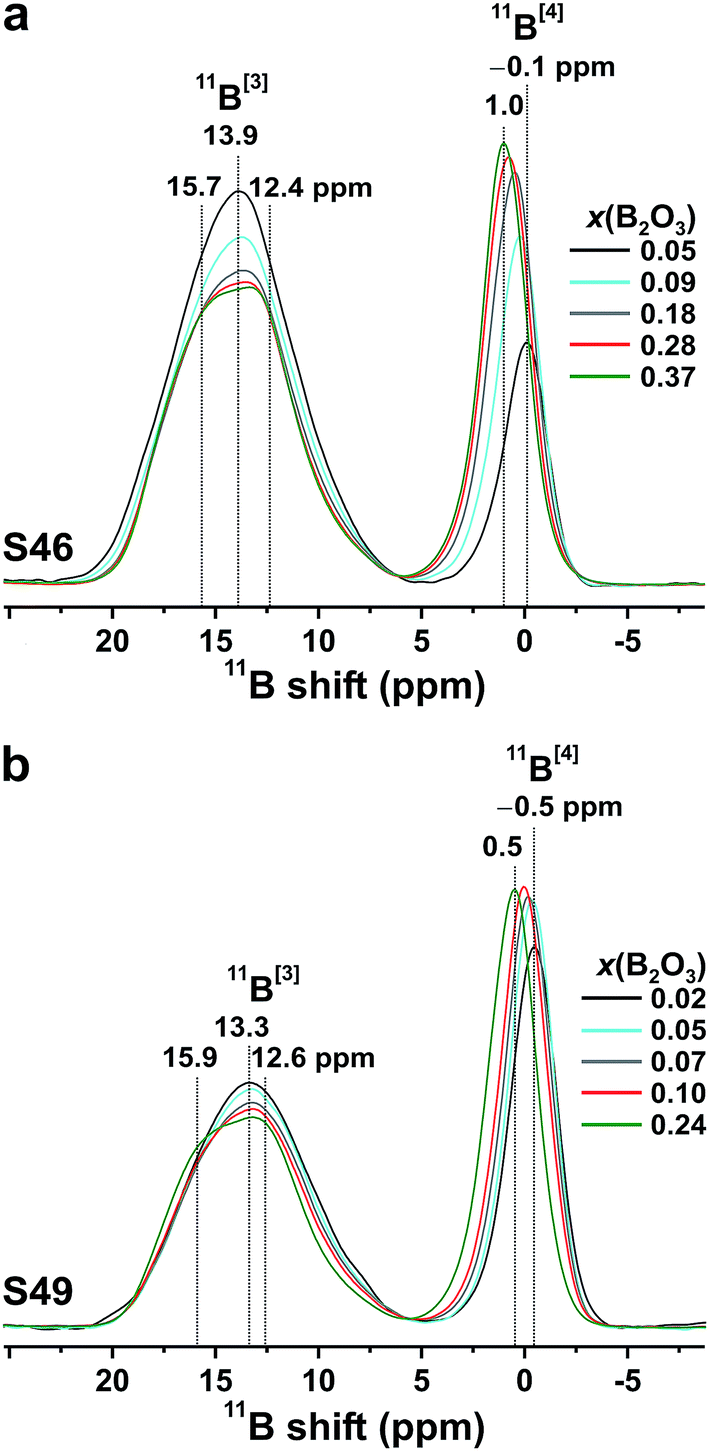 | ||
| Fig. 3 11B MAS NMR spectra obtained at 14.1 T from the (a) S46 and (b) S49 glasses for the as-indicated x(B2O3) values. The total integrated signal intensity of each NMR spectrum is normalized to unity. | ||
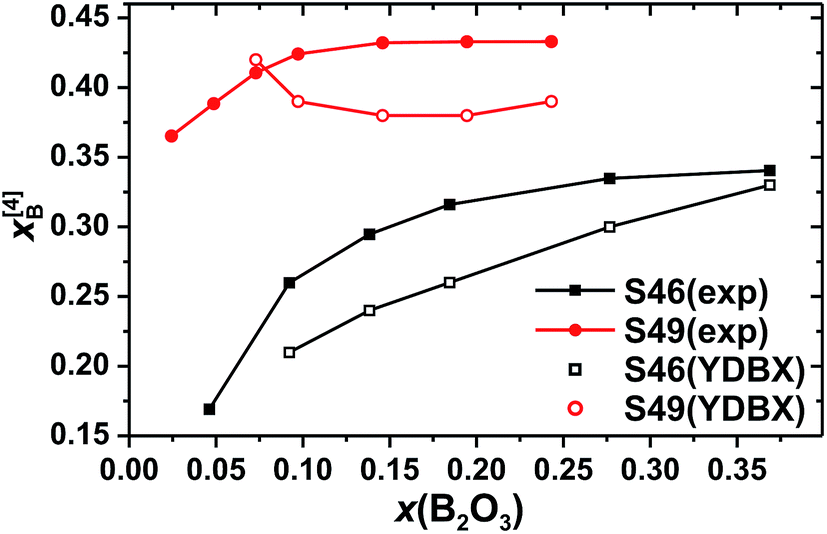 | ||
| Fig. 4 The fractional population of B[4] groups (out of the total B speciation) plotted against the molar fraction of B2O3 for the S46 (solid squares) and S49 (solid circles) glass series. Open symbols represent predictions from the Yun–Dell–Bray–Xiao (YDBX) model.38 | ||
4.3 11B NMR
![[C with combining macron]](https://www.rsc.org/images/entities/i_char_0043_0304.gif) Q, with CQ = e2qQ/h) owing to the higher symmetry of the charge distribution around the 11B nucleus.37,38,46,47 The following general trends are observed in Fig. 3 when SiO2 is substituted by B2O3 at a constant network modifier content: (i) the intensity of the 11B[4] resonance increases due to B[3] → B[4] conversions,37,38,45,47 while its position displaces monotonically towards higher shifts. As analyzed further in Section 5.1, this reflects a concurrent decrease in the number of Si atoms in the second coordination shell of 11B.44,45,49–52 (ii) The 11B[3] resonance alters from an essentially featureless peak, centered at 13.9 ppm (S46) and 13.3 ppm (S49) from the Si-rich glass compositions, to a broad signal revealing two maxima ≈15.8 ppm and ≈12.5 ppm; see Fig. 3. This suggests that the 11B NMR signals in the ≈8–20 ppm spectral region stem from at least two distinct 11BO3 environments in all Si-dominated glasses, as discussed further below.
Q, with CQ = e2qQ/h) owing to the higher symmetry of the charge distribution around the 11B nucleus.37,38,46,47 The following general trends are observed in Fig. 3 when SiO2 is substituted by B2O3 at a constant network modifier content: (i) the intensity of the 11B[4] resonance increases due to B[3] → B[4] conversions,37,38,45,47 while its position displaces monotonically towards higher shifts. As analyzed further in Section 5.1, this reflects a concurrent decrease in the number of Si atoms in the second coordination shell of 11B.44,45,49–52 (ii) The 11B[3] resonance alters from an essentially featureless peak, centered at 13.9 ppm (S46) and 13.3 ppm (S49) from the Si-rich glass compositions, to a broad signal revealing two maxima ≈15.8 ppm and ≈12.5 ppm; see Fig. 3. This suggests that the 11B NMR signals in the ≈8–20 ppm spectral region stem from at least two distinct 11BO3 environments in all Si-dominated glasses, as discussed further below.The {x[3]B, x[4]B} populations were determined from the integrated CT NMR signal intensities of the respective {11BO3, 11BO4} groups, including the weak first-order 11BO3 CT spinning sidebands. Each 11BO4 intensity was corrected for the contributions of the ST centerband (which overlap with the main CT resonance) according to the procedure of Massiot et al.;68 the correction yielded 0.02–0.03 lower x[4]B values relative to the corresponding uncorrected populations. The results are listed in Table 3, together with the mean isotropic chemical shifts and average quadrupolar products Qη of the 11BO4 moieties (CQη relates to CQ according to 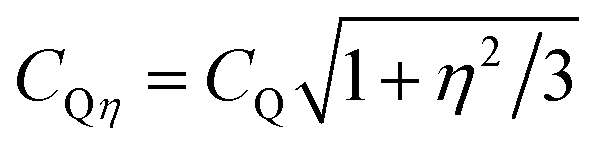 , where η is the asymmetry parameter of the quadrupolar tensor1–3). These parameters were obtained by analyzing the CT/ST signals of the 11B MAS NMR spectra as described in ref. 69. We stress that all {
, where η is the asymmetry parameter of the quadrupolar tensor1–3). These parameters were obtained by analyzing the CT/ST signals of the 11B MAS NMR spectra as described in ref. 69. We stress that all {![[small delta, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c5.gif) iso, Qη} parameters listed in Table 3 are mean values of (up to) three co-existing 11BO4 environments with variable numbers of Si/B[3] neighbors (see Section 5.1), whose individual NMR parameters cannot be determined.
iso, Qη} parameters listed in Table 3 are mean values of (up to) three co-existing 11BO4 environments with variable numbers of Si/B[3] neighbors (see Section 5.1), whose individual NMR parameters cannot be determined.
| Label | B[3] | B[4] | Populationsb | BO3 populationsc | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| δCG (ppm) | iso (ppm) |
Qη (MHz) |
δCG (ppm) | iso (ppm) |
Qη (MHz) |
x[3]B | x[4]B | x[3]B(0Si) | x[3]B(1Si) | |
| a δCG(B[3]) and δCG(B[4]) with uncertainties of ±0.08 ppm and ±0.04 ppm, respectively, correspond to the center-of-gravity shifts of the 11BO3 and 11BO4 resonances in the MAS NMR spectra. All other NMR parameters for the 11BO3 and 11BO4 groups were obtained from 3QMAS and MAS 11B NMR spectra, respectively, with uncertainties as follows: iso(B[3])±0.20 ppm; iso(B[4])±0.06 ppm; Qη(B[3])±0.10 MHz; Qη(B[4])±0.10 MHz.b The fractional populations (uncertainty ±0.01) were obtained by integrating the MAS NMR intensities and then correcting for the satellite-transition contributions to the CT centerband peak.68c Relative populations of BO3 groups without [x[3]B(0Si)] and with one [x[3]B(1Si)] bond to Si (uncertainty ±0.03); they are normalized according to x[3]B(0Si) + x[3]B(1Si) = 1. |
||||||||||
| S462.6(5) | 14.00 | −0.04 | 0.12 | 0.39 | 0.831 | 0.169 | ||||
| S462.6(9) | 13.98 | 18.6 | 2.72 | 0.31 | 0.43 | 0.35 | 0.740 | 0.260 | 0.76 | 0.24 |
| S462.6(14) | 13.98 | 0.49 | 0.59 | 0.36 | 0.705 | 0.295 | ||||
| S462.6(18) | 13.96 | 18.7 | 2.74 | 0.58 | 0.72 | 0.35 | 0.684 | 0.316 | 0.86 | 0.14 |
| S462.6(28) | 14.01 | 0.83 | 0.94 | 0.38 | 0.665 | 0.335 | ||||
| S462.6(37) | 14.02 | 18.8 | 2.70 | 1.02 | 1.12 | 0.39 | 0.660 | 0.340 | 1.00 | 0.00 |
| S494.0(2) | 13.21 | −0.47 | −0.31 | 0.37 | 0.635 | 0.365 | ||||
| S494.0(5) | 13.22 | −0.29 | −0.19 | 0.35 | 0.612 | 0.388 | ||||
| S494.0(7) | 13.29 | −0.15 | −0.01 | 0.36 | 0.589 | 0.411 | ||||
| S494.0(10) | 13.40 | 18.2 | 2.70 | 0.09 | 0.20 | 0.37 | 0.576 | 0.424 | 0.70 | 0.30 |
| S494.0(15) | 13.62 | 0.39 | 0.49 | 0.37 | 0.568 | 0.432 | ||||
| S494.0(19) | 13.65 | 0.51 | 0.61 | 0.38 | 0.567 | 0.433 | ||||
| S494.0(24) | 13.72 | 18.4 | 2.72 | 0.59 | 0.72 | 0.40 | 0.567 | 0.433 | 0.83 | 0.17 |
Fig. 4 plots the B[4] fractional population (x[4]B) against x(B2O3). Both S46/S49 series reveal a strong growth of x[4]B when x(B2O3) is increased at low substitution levels [x(B2O3) ≲ 0.07]. When the amount of B is elevated further, x[4]B tends towards asymptotic values of ≈0.34 and ≈0.43 for the S46 and S49 glass families, respectively. However, while the plateau is reached at already ≈30% B2O3 for SiO2 substitution of the S49 glasses, the B[4] population manifests a slow increase throughout the entire range of B2O3 contents in the S46 series. Noteworthy, the increase of x[4]B (by ≈0.17) across the S46 glasses is more than twice that (≈0.07) observed for the S49 members. As expected from the 11B[4] peak intensities of Fig. 3, the overall Si-richer S49 glasses exhibit higher relative B[4] populations than their S46 counterparts. While this trend is predicted by the YDBX model,37,38 it accounts only qualitatively for the de facto observed alterations of the experimental B[4] populations for variable amounts of B; see Fig. 4.
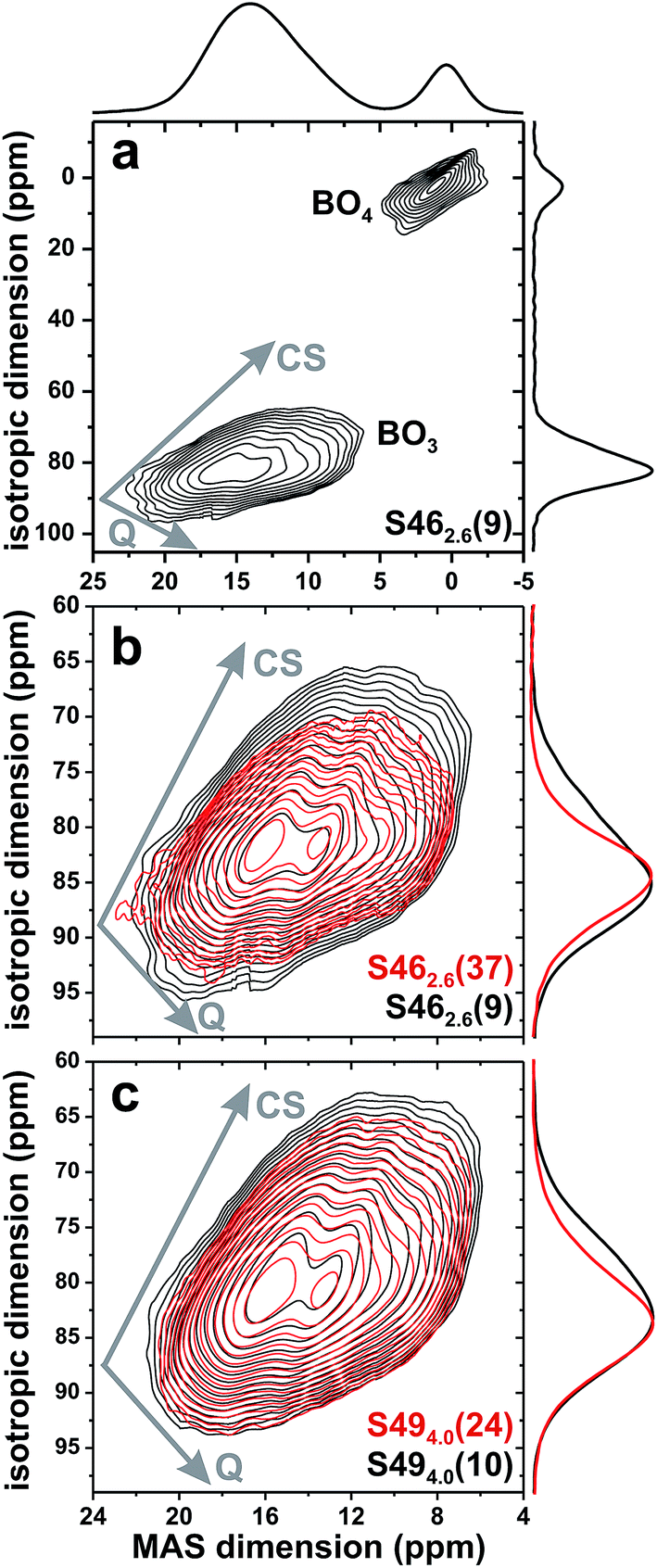 | ||
| Fig. 5 (a) 3QMAS 11B NMR spectrum recorded at 14.1 T from the S462.6(9) glass. (b and c) Zooms around the 11B[3] signal region of 3QMAS NMR spectra obtained from (b) S462.6(9) (black contours); S462.6(37) (red contours), and (c) S494.0(10) (black contours); S494.0(24) (red contours). The arrows mark the directions of the resonance spreads stemming from distributions of isotropic chemical shifts (marked by “CS”) and quadrupolar coupling constants (labeled “Q”). | ||
Fig. 5(b) and (c) shows zoomed areas around the 11B[3] 2D NMR ridge, where pairs of spectra from glasses with low and high amounts of B are superimposed from the (b) S46 and (c) S49 glass series. Each Si-dominated glass reveals a higher NMR signal-dispersion towards lower shifts relative to its B-richer counterparts: it is manifested by an enhanced NMR intensity ≈74 ppm in the corresponding projection along the isotropic dimension and suggests that the net 11B[3] resonance stems from two (or more) distinct BO3 structural environments. This is most transparent for the two S46 samples that exhibit the largest span of B contents; see Fig. 5(b).
The correlation between the signal intensity ≈74 ppm and the relative SiO2 content of the glass is evident from the isotropic projections shown in the left panel of Fig. 6. The 11B NMR peakshape from the almost Si-free S462.6(37) glass fitted well to a single Gaussian peak centered ≈82 ppm [Fig. 6(a)], indicating that its structure comprises primarily one type of BO3 structural group. Given the domination of B relative to Si in the glass network (xB/xSi = 8.00; see Table 1), we attribute it to a BO3 environment participating in boroxol rings50–52,55 and devoid of bonds to Si atoms; this moiety is consequently labeled B[3](0Si). The lower-ppm signal grows for increasing SiO2 content, indicating it is associated with a “non-ring” BO3 environment connected to one SiO4 group, which we denote by 11B[3](1Si).
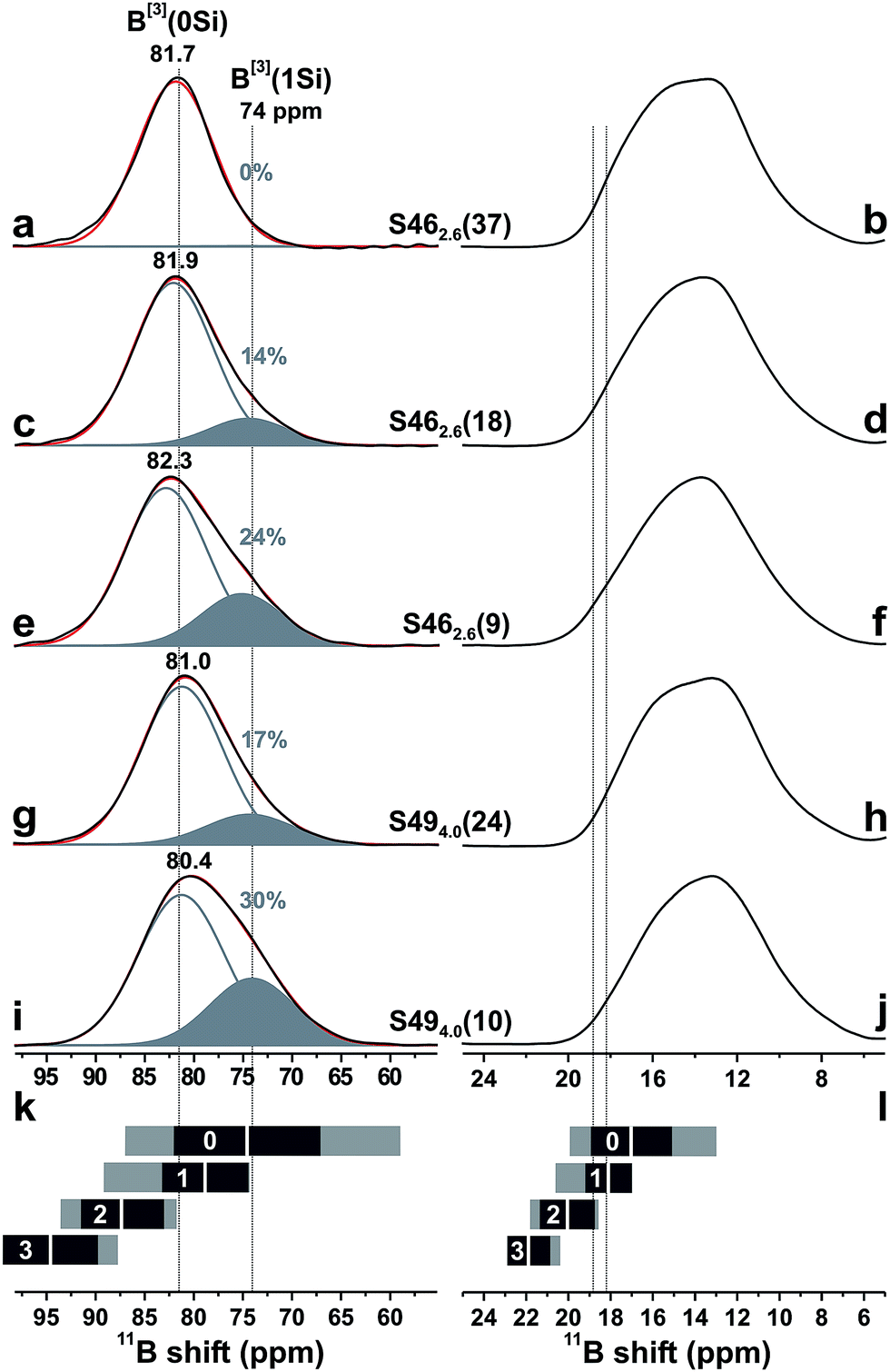 | ||
| Fig. 6 Projections along the isotropic dimension of 3QMAS 11B NMR spectra (left panel) and MAS spectra (right panel) from the as-indicated S46 and S49 glasses. The black and red traces represent experimental and best-fit peakshapes, respectively: they comprise two signal components at ≈80–82 ppm (gray lines; 11B[3](0Si) groups in boroxol rings) and ≈74 ppm (solid areas; 11B[3](1Si) moieties). Gray numbers specify the fractional populations (in %) of the B[3](1Si) groups. (k) Ranges of center-of-gravity (CG) shifts along the isotropic projection for 11B[3] moieties with 0, 1, 2, and 3 NBO ions, compiled from literature data:51,55,56,72–79 the white bar marks CG, whereas the gray and black rectangles depict the total shift-span and CG ± σ, respectively. (l) As in (k), but illustrating the range of {iso}, which is to be compared with the span of experimentally observed extreme iso-values (dotted lines) at 18.2 ppm and 18.8 ppm; see Table 3. | ||
The relative 11B[3](0Si) [“ring”] and 11B[3](1Si) [“non-ring”] contributions were extracted by deconvoluting each NMR spectrum into two components; see Fig. 6. The variable 11B[3](0Si) and 11B[3](1Si) peak intensities are responsible for the lineshape alterations of the MAS spectra in Fig. 3 when the xB/xSi ratio of the glass is changed. The B[3](1Si) population grows up to 24% when x(B2O3) is decreased from 0.37 to 0.09 in the S46 series; it amounts to 30% out of the total B[3] speciation in the Si-richest glass [S494.0(10)] that was examined by 3QMAS 11B NMR; see Fig. 6(i). As follows by comparing the results for S462.6(9) and S494.0(10) that feature a constant B2O3 for SiO2 substitution degree of 20%, the B[3](1Si) contribution is larger for the Si-richer BPS glass with lower NBO content. These trends of growing “non-ring” fraction for increasing (decreasing) amount of Si (NBO) accord with the results of Du and Stebbins for alkali-poor borosilicate glasses.50,51
5 Discussion
5.1 BO4 environments
Given that the total Na+/Ca2+ content remains constant within each S46 and S49 series, the 11B[4] resonance-displacement may be accounted for by a fixed set of groups, {11B[4](mSi)}, each featuring m bonds to Si atoms (and thereby 4 − m bonds to B[3]) and resonating at the CG shift δ[4]B(mSi). Several investigations report a propensity for BO4 groups to connect to SiO4 tetrahedra rather than the planar BO3 moieties,37,38,50–52 whereas direct B[4]–O–B[4] linkages are often assumed to be absent in the borosilicate structure.1–3,37,38,50–52 Hence, B[4](4Si) environments are expected to dominate the BO4 speciation in the Si-richest BPS glasses. Yet, considering the wide ranges of xB/xSi ratios sampled across the S46/S49 glass series (notably so for the S46 set; see Table 1), more than two structural moieties should co-exist: with the constraints of combining physically reasonable {δ[4]B(mSi)} values with decent “best fits”, three {11B[4](4Si), 11B[4](3Si), 11B[4](2Si)} groups were selected and each 11BO4 NMR signal of Fig. 3 was deconvoluted into Gaussian-shaped peaks centered at the nearly constant (±0.1 ppm) shift-values {−0.45, 0.65, 1.85} ppm for all glasses. We stress that these NMR spectra deconvolutions are somewhat arbitrary because the set of precise {δ[4]B(mSi)} values are a priori unknown. The proposed shift alteration associated with a B[4](mSi) → B[4]([m − 1]Si) conversion varies between various studies, where (for instance) shift differences of 0.7–1.0 ppm,47,71 and 1.7–2.0 ppm (ref. 51 and 52) are suggested.
Fig. 7 presents the set {x[4]B(mSi)} of best-fit fractional populations plotted against each ratio
| yB = xB/(xB + xSi), | (3) |
| y[3]B = xBx[3]B/(xBx[3]B + xSi), | (4) |
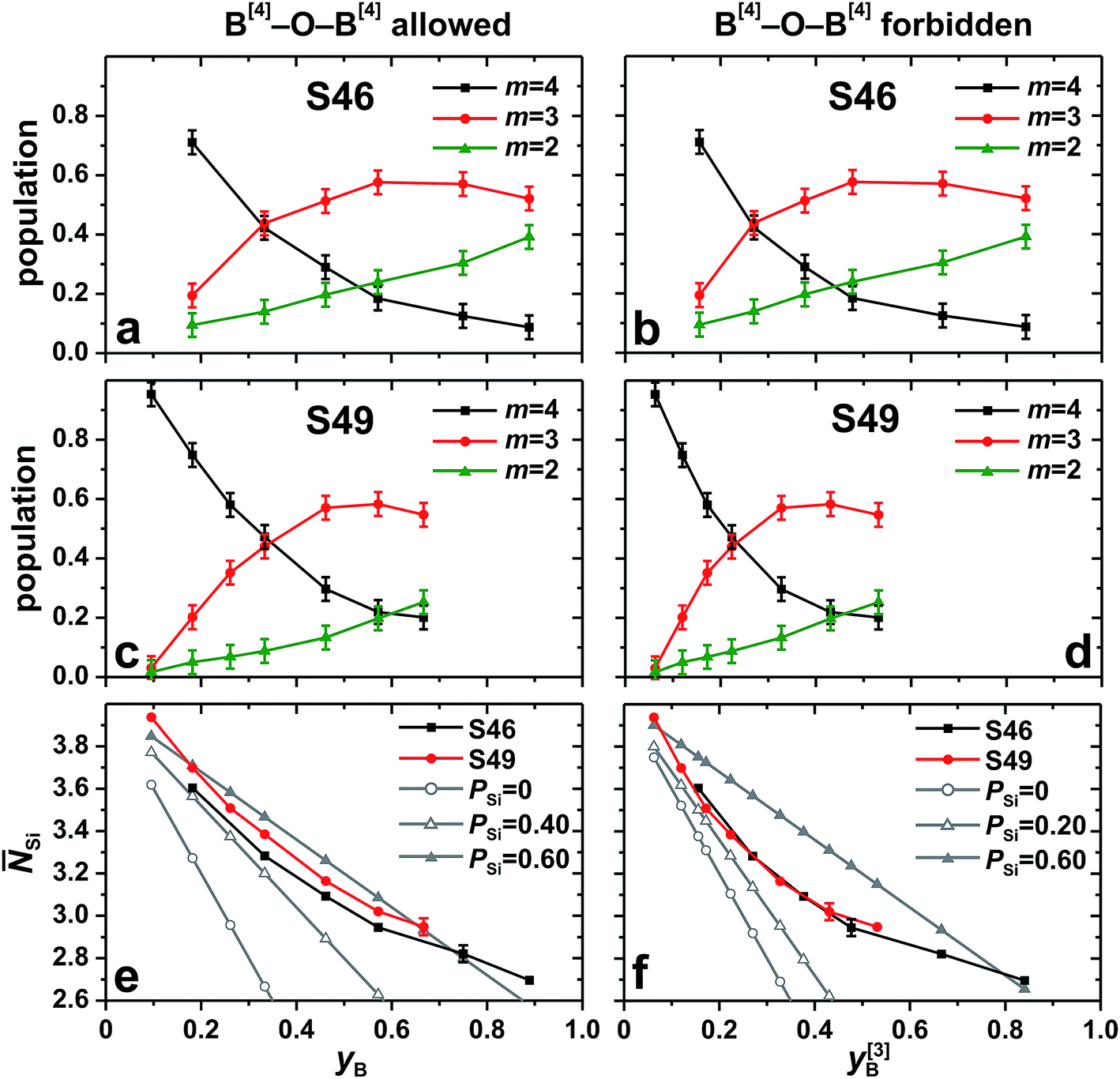 | ||
| Fig. 7 (a–d) NMR-derived best-fit populations [x[4]B(mSi)] of BO4 groups with m B–O–Si bonds observed from the series of (a, b) S46 and (c, d) S49 glasses. The data are plotted against the fractions (a, c, e) yB = xB/(xB + xSi) and (b, d, f) y[3]B = xBx[3]B/(xBx[3]B + xSi). yB and y[3]B are relevant for the scenarios of unrestricted B[4]–O–B[3]/B[4]/Si bond formation and B[4]–O–B[4] avoidance, respectively. (e, f) Average number of Si atoms per BO4 group (Si) obtained from the fractional populations in (a, c) and (b, d), respectively. The experimental data are compared with the as-indicated statistical models, where PSi = 0 corresponds to a random bond formation between B[4] and atoms from the set (e) {Si, B[3], B[4]} or (f) {Si, B[3]}, whereas PSi > 0 implies a preference for B[4]–O–Si linkages [see eqn (5)]. To improve visualization in (e, f), the error bars (±0.04) are generally suppressed. | ||
 . The corresponding average number of B atoms (B) is given from the constraint B = 4 − Si, where “B” represents the total B speciation in Fig. 7(e) but solely the B[3] coordinations in (f). The mean number of Si neighbors around the BO4 groups decreases from Si = 3.62 in the Si-rich S462.6(5) structure to Si = 2.70 in the B-rich S462.3(37) glass. The corresponding data observed for the S49 series are Si = 3.94 [S494.0(2)] and Si = 2.95 [S494.0(24)]. These overall high Si-values underscore the preference for B[4]–O–Si contacts relative to B[4]–O–B. This inference is obvious when contrasting the experimentally observed {Si} data with the scenario of random distributions of either the Si/B[3] species [Fig. 7(f)] or of all Si/B[3]/B[4] moieties [Fig. 7(e)] around the BO4 groups. Besides that the statistical (binomial) distribution reveal a markedly faster drop of Si than that observed experimentally, it leads to substantial {x[4]B(0Si), x[4]B(1Si)} populations (data not shown) that are incommensurate with the experimental observation of (up to) three co-existing B[4](mSi) structural groups.
. The corresponding average number of B atoms (B) is given from the constraint B = 4 − Si, where “B” represents the total B speciation in Fig. 7(e) but solely the B[3] coordinations in (f). The mean number of Si neighbors around the BO4 groups decreases from Si = 3.62 in the Si-rich S462.6(5) structure to Si = 2.70 in the B-rich S462.3(37) glass. The corresponding data observed for the S49 series are Si = 3.94 [S494.0(2)] and Si = 2.95 [S494.0(24)]. These overall high Si-values underscore the preference for B[4]–O–Si contacts relative to B[4]–O–B. This inference is obvious when contrasting the experimentally observed {Si} data with the scenario of random distributions of either the Si/B[3] species [Fig. 7(f)] or of all Si/B[3]/B[4] moieties [Fig. 7(e)] around the BO4 groups. Besides that the statistical (binomial) distribution reveal a markedly faster drop of Si than that observed experimentally, it leads to substantial {x[4]B(0Si), x[4]B(1Si)} populations (data not shown) that are incommensurate with the experimental observation of (up to) three co-existing B[4](mSi) structural groups.To asses the degree of preference for B[4]–O–Si bond formation relative to B[4]–O–B, we employed the procedure introduced recently by Mathew et al.34 It applies to a scenario where two distinct atom species V and W may be coordinated by U: when there is a preference for U–V contacts, the preference factor PV—where the subscript “V” denotes the preferred species—encodes the deviation from a statistical V/W distribution around U (for which PV = PW = 0), where PV = 1 represents the case of sole U–V bonding. Within this framework, the preference factor PSi dictates the average number of Si (Si) and B (B) atoms around the B[4] coordinations as follows:34
|
Si = 4[PSi + ySi(1 − PSi)],
| (5) |
|
B = 4yB(1 − PSi) for equal B[3]/B[4] probabilities
| (6) |
| = 4y[3]B(1 − PSi) for B[4]–O–B[4] avoidance, | (7) |
B.
For each glass, the preference for B[4]–O–Si bonds was calculated from the experimental data of Fig. 7(a)–(d) by using eqn (5) and solving for PSi. Onwards disregarding the result for the Si-richest S494.0(2) glass that is considered to be an outlier, no striking differences are observed if B[4]–O–B[4] linkages would be absent or present, with the respective {PSi} data typically scattering around the values 0.45 and 0.55. Consequently, the scenario of B[4]–O–B[4] avoidance is associated with a somewhat less pronounced propensity for B[4]–O–Si bond formation; this is evident from Fig. 7(e) and (f), where the experimental Si values of nearly all members from the S46 and S49 families are bracketed by the results stemming from distribution models associated with PSi = 0.60 and the corresponding lower limit of PSi = 0.40 if B[4]–O–B[4] bonds are allowed [Fig. 7(e)] and PSi = 0.20 when B[4]–O–B[4] links are absent [Fig. 7(f)].
To conclude, the BO4 moieties in both S46 and S49 glass families display a clear preference for connecting to SiO4 tetrahedra rather than to BO3/BO4 groups. The observed typical values of PSi ≈ 0.5 are equivalent to a Si/B distribution where Si atoms first occupy two out of four neighboring positions around each BO4 tetrahedron, whereas Si and B atoms are statistically distributed around the remaining two tetrahedral corners. However, considering the uncertainties and possible systematic errors of the experimental Si data, the precise values should not be taken too literally and further studies are required to better confine the relative preferences, as well as if “B[4] avoidance” applies to modifier-rich BPS glass networks.
5.2 NBO partitioning among SiO4 and BO3 groups
The affinity for coordinating NBO ions among the three {P, Si, B} network formers decreases in the order P ≫ Si > B[3] ≫ B[4].3,40 Because B[4]–NBO bonds are assumed to be absent3,40 and the NBO accommodation among the Q0P (PO43−) and Q1P (PO3.52−) moieties are known accurately (see Table 2), we henceforth focus on the NBO partitioning among the SiO4 and BO3 groups.The total NBO population available for depolymerizing the borate/silicate networks may be estimated for each glass after accounting for the consumption of Na+/Ca2+ cations by the phosphate groups [see eqn (1)] and the [BO4]− tetrahedra. By using the average silicate network connectivity (SiBO) available from the Q1P population (Section 4.2), the mean numbers of BO (BBO) and NBO (BNBO) species at the BO3 groups may be estimated. The S46 and S49 glasses exhibit relatively narrow SiNBO ranges of 1.76–1.89 and 1.27–1.46, respectively, yielding the corresponding approximate BNBO-values of 1 and 0.7–0.8. Hence, the {BNBO, SiNBO} parameter-pair only alters slightly across each S46/S49 glass family when the xB/xSi ratio varies. It is instructive to compare the results conveying the relative propensities for Si–NBO and B[3]–NBO bond formation between the two series, which reveal the pairs {BNBO, SiNBO} ≈ {1, 2} for S46 and {BNBO, SiNBO} ≈ {0.75, 1.5} for S49. For the S46 glass members, this implies roughly two and one NBO species per SiO4 and BO3 group, respectively. In contrast, the more polymerized S49 networks involve SiO4 tetrahedra that on the average coordinate ≈1.5 NBO (consistent with similar Q2Si and Q3Si populations; see Fig. 2), while ≈25% of the {BO3} ensemble is devoid of NBO species.
Given that the S46 glass networks involve an NBO distribution peaked around ≈1 NBO per BO3 moiety, whereas those of the S49 structures comprise BO3 groups that accommodate either 0 or 1 NBO, one may ask if our 11B NMR data of Fig. 3 and 6 may resolve these resonances. The shift-spans compiled in Fig. 6(k) from literature data51,55,56,72–79 (relevant for the peak maxima along the isotropic projection of 3QMAS spectra) suggest an overall minor deshielding of the 11BO3 environments for an increasing number of NBO anions, but also manifests strongly overlapping shift ranges from 11BO3 moieties with 0 and 1 NBO ions. Hence, these resonances cannot be separated with the 3QMAS NMR spectra resolution offered from our glasses. Nevertheless, significant contributions from 11BO3 groups with two or three NBO species may be excluded [Fig. 6(k)], in full accordance with our estimates above and further corroborated by the “typical” isotropic chemical-shift ranges displayed in Fig. 6(l) that are relevant for the 11B MAS NMR spectra of the right panel of Fig. 6.
5.3 BPS structural model
The networks of the parent B-free S462.6(0) and S494.0(0) glasses comprise mainly inter-connected Q2Si and {Q2Si, Q3Si} silicate groups, respectively (see Fig. 2). A slight increase in SiBO by up to ≈0.2 results when B is introduced, because some NBO ions relocate to the BO3 groups, which on the average accommodate ≈1 and ≈0.7–0.8 NBO species in the B-bearing S46 and S49 glasses, respectively. Yet, the {QnSi} speciation is expected to remain overall intact, except that Si–O–Si bonds are gradually replaced by Si–O–B[4] (major) and Si–O–B[3] (minor) linkages in the borosilicate network. The tetrahedral SiO4 and BO4 species strongly prefer to interconnect. This implies that B[4](4Si) groups solely involving bonds to Si account for >70% of all BO4 units in the Si-richest S494.0(2), S494.0(5), and S462.6(5) glasses; see Fig. 7. The average number of bonds to Si remains ≳3 until ≈40% of the SiO2 reservoir is replaced by B2O3. Yet, a relatively high mean number Si ≈ 2.7 of B[4]–O–Si bonds persists even in the B-richest S462.6(37) structure. The estimated preference factor PSi ≈ 0.5 is equivalent to a scenario with two Si neighbors around each BO4 tetrahedron, while the remaining two positions are filled statistically by Si and B[3] atoms according to their relative abundances in the glass. The B[3] coordinations prefer connecting to other species according to B[3] > B[4] > Si. Hence, despite that the total number of B[3]–O–B[4] bonds increases concurrently with the B2O3 content at the expense of the B[3](1Si) [i.e. “non-ring”] fraction, the B[3](0Si) [“ring”] groups mainly associate with themselves as boroxol rings, regardless of the precise xB/xSi ratio and total NBO content of the BPS structure.
The relative proportions of these borate/borosilicate domains depends on the xB/xSi ratio of the glass. For instance, the Si-rich S462.6(5) structure associated with xB/xSi = 0.22 and x[4]B = 0.17 (see Tables 1 and 3) is dominated by a silicate network of Q2Si groups cross-linked by B[4](4Si) species. This Si-rich borosilicate network co-exist with a domain of boroxol rings featuring ≈1 NBO per BO3 moiety. The two networks are merged by B[4](3Si) and B[3](1Si) groups. When the B2O3 content is increased, the number of BO4 groups increases in the borosilicate network, while B[4](3Si), B[4](2Si)—and to a lesser extent B[3](1Si)—species constitute bridges to the boroxol rings. The B-richest S462.6(37) structure comprises mainly boroxol rings that are cross-linked by B[4](3Si) and B[4](2Si) groups to a borosilicate network dominated by BO4 groups; the latter is overall more condensed than those of the Si-rich S46 glasses, due to the absence of NBO species at the BO4 tetrahedra and because the Q2Si groups are minor components (xB/xSi = 8.00). Since the BO3 groups now grossly outnumber the SiO4 tetrahedra, most of the NBO reservoir resides at the boroxol rings (Section 5.2).
5.4 Implications for glass dissolution
Here we discuss our structural model in conjunction with general glass degradation trends reported in the literature for borosilicate/BPS glasses. We identify two structural descriptors that we expect to primarily control the dissolution rate of BPS glasses in aqueous solutions, and highlight some remaining structural questions that must be addressed for achieving bioactive BPS glasses with tailored solubility in (simulated) body fluids.On their immersion in aqueous solutions, borate-based BGs are claimed to (A) degrade faster and (B) convert more completely to HCA relative to their silicate-based counterparts.4–6,11,12 However, few (if any) direct experimental comparisons of the HCA formation among borate-bearing/free BGs are reported. The hitherto lacking detailed structural understanding of BPS glasses hampers a rational design for tuning the glass degradability (“solubility”) in aqueous media.
Concerning (B), it remains unclear if the HCA formation rate or amount thereof formed are obviously higher than those observed from highly bioactive silicate-based BGs such as the “45S5” composition,15 i.e., S462.6(0). As commented by Lepry and Nazhat,14 essentially all in vitro evaluations of HCA formation from B-bearing BGs employed solutions with 20–250 times higher phosphate concentrations than human plasma or its acellular simulated body fluid (SBF)83 counterpart that has become the standard medium for in vitro testing. For a melt-prepared B-based 45S5 composition4,5 (S462.6(46) in our notation), an induction time of 3 days for HCA formation in SBF was reported,14 which is significantly longer than that ≲12 h (ref. 84) observed for the original 45S5 formulation. More direct in vitro comparisons between silicate/borate-based BGs in SBF are required to evaluate their relative merits for HCA formation.
We next consider the glass degradability in aqueous media, i.e., (A). For (phospho)silicate glasses, it is well-known that a decrease in the silicate network connectivity (SiBO) accelerates the glass dissolution.20,23,42 Solubility data are not gathered for our BPS glasses, but one expects from the more condensed S494.0(0) network (SiBO = 2.54) that it degrades slower than its S462.6(0) counterpart (SiBO = 2.11). The literature inarguably shows that the incorporation of B into a silicate-based glass increases its dissolution in aqueous media,19,20 as well as that bioactive BPS glasses dissolve more rapidly than their B-free counterparts.4–9 However, that same literature on BG applications is vague regarding the underlying reasons: by assuming that B is exclusively present as BO3 groups in the glass structure, Yao et al. attributed the comparatively faster degradation of borate/borosilicate glasses to the lower connectivity of the B[3] coordinations relative to Si[4].5 Yet, the silicate network connectivity of bioactive silicate-based glasses remains SiBO < 3 (ref. 23, 41 and 42) and is consequently lower than that of vitreous B2O3. Moreover, as shown herein, the networks of Na/Ca-modified BPS glasses also comprise fully polymerized—and thereby highly cross-linking—BO4 tetrahedra. The higher degradability of B-bearing phosphosilicate glasses merely stems from the higher hydrolysis rate of B–O bonds relative to Si–O, with the reactivity increasing in the order19,20
| Si–O–Si < Si–O–B[4] < B[3]–O–B[3]. | (8) |
This reactivity order coupled with the progressive replacements of Si–O–Si by Si–O–B and (particularly) B–O–B bonds rationalize the elevated glass dissolution observed4,5 when B replaces Si in the (boro)silicate networks. Moreover, the combined effects from the higher cross-linking of B[4] coordinations and a slower hydrolysis of the B[4]–O–Si bonds relative to the B[3]–O–B[3] linkages19,20 make the borosilicate network portions of the BPS glass more durable. At a constant cation composition and total NBO content, it follows that the higher the x[4]B fraction in the BG structure, the more resistant to aqueous attack the glass structure becomes. Hence, the parameter-pair {SiBO, x[4]B} should primarily govern the solubility of BPS glasses, where the following trends are predicted for the S46/S49 members: (i) at a fixed xB/xSi ratio, the lower {SiBO, x[4]B} values of an S46-based glass implies a faster degradation than its S49 analog. (ii) However, relative to the S46 networks, the S49-based counterparts should exhibit less variations in solubility for increasing B2O3 substitution degree, since the alteration of the B[4] fraction is smaller among the S49 members (see Table 3). We note that all arguments above draw from well-established dissolution trends of (P-free) borate/borosilicate glasses,19,20 which become applicable to the present BPS glasses with low P contents thanks to the herein established BPS structural model of borate/borosilicate networks surrounded by orthophosphate ions (see Sections 4.1 and 5.3.1). Hence, when ignoring the small Q1P population, any given BPS glass network is an approximation of a borosilicate counterpart, as argued in Section 2.2.
Noteworthy, x[4]B increases concurrently with x(SiO2) at constant modifier content in the glass; see Table 3. Hence, lowering the SiO2 content improves the solubility because both the numbers of Si–O–Si bonds and network-condensing BO4 moieties are reduced. The YDBX model37,38 predicts that a further increase in the total Na+/Ca2+ content of these modifier-rich bioactive BPS glasses drives B[4] → B[3] conversions at a constant xB/xSi ratio. Consequently, increasing the modifier content of a borosilicate/BPS glass will accelerate its degradability because either/both of the {SiBO, x[4]B} parameters is/are decreased. Future work will need to settle which structural role the surplus modifiers predominantly assumes—as charge compensators of BO4 groups or of NBO ions? Such insight combined with the herein reported near invariance of the silicate network polymerization when Si is replaced by B at a constant modifier content (Section 4.2) should allow for tuning each {SiBO, x[4]B} parameter almost independently for a given {xB, xSi, xP} glass composition. We stress that our proposed role of {SiBO, x[4]B} to (primarily) govern the BPS glass dissolution is independent on how these values were arranged; while SiBO and x[4]B depend foremost on the BPS glass composition, particularly the B speciation may display a weak dependence on the thermal history of the glass.
6 Conclusions
We have proposed a structural model for modifier-rich Na2O–CaO–B2O3–SiO2–P2O5 glasses with relatively low P contents [x(P2O5) ≤ 0.04] and variable x(SiO2)/x(B2O3) ratios. The following structural alterations are observed when SiO2 is gradually replaced by B2O3 at a constant glass-modifier content:(i) The phosphate speciation and the average silicate network polymerization only change marginally, where the minor increase of the Q1P population (reflecting the extents of Si–O–P bonding) stems from the concurrent silicate-network condensation.33 Both these trends originate from (ii) a redistribution of NBO species from the SiO4 tetrahedra to the BO3 groups, which on the average accommodate 0.7–1 NBO ions per B atom. (iii) Notwithstanding that B[3] coordinations dominate the B speciation throughout all glasses, a progressive B[3] → B[4] conversion is observed for increasing x(B2O3). Yet, regardless of the B2O3 content, the more condensed and Si-rich S49 glass networks manifest consistently higher fractional populations of BO4 tetrahedra (37–43%) than their S46 counterparts (17–34%). The x[4]B fraction in the S49 glasses reaches a plateau at ≈43% already when 30% of the SiO2 reservoir is replaced by B2O3, whereas minor transformations from trigonal to tetrahedral B coordinations are observed throughout the entire range of B-for-Si substitutions in the S46 series.
The medium-range BPS glass structure is mainly governed by the strong preferences for B[3]–O–B[3] and B[4]–O–Si bonding scenarios relative to other options, such as B[3]–O–B[4] and B[3]–O–Si. Along previous reports on borosilicate glasses of low modifier content,50–52 substantial amounts of B[3]–O–Si contacts are only observed in Si-rich BPS networks, whereas they are sparse in structures exhibiting comparable amounts of B and Si. From these bonding preferences, we inferred a BPS structural model that comprises (sub)nm-sized “domains” of a borate network (BO3 groups in boroxol rings) interlinked with a borosilicate network through B[4](3Si), B[4](2Si), and B[3](1Si) moieties.
We proposed that the parameter-pair {SiBO, x[4]B} primarily dictates the solubility of alkali/alkaline-earth bearing BPS glasses, whose degradation should decrease concurrently with an increase in either the silicate network polymerization or the B[4] population. The improved insight about the alterations of these parameters when Si is replaced by B gained from our study should allow for tailoring the glass solubility in (simulated) body fluids. Moreover, for biomedical applications, a high amount of readily leached orthophosphate ions is believed to improve the HCA formation.23,33,36 We are currently exploring the glass-formation limits of P-richer BPS glasses to verify that the beneficial structural feature of a PO43−-dominated phosphate speciation remains also in this composition regime, as well as for locating the permissible range of x(SiO2)/x(B2O3) and x(Na2O)/x(CaO) molar ratios that yield homogeneous glasses. Further work is required to verify the overall validity of the herein proposed borate/borosilicate BPS network organization, which was conjectured from standard 29Si, 31P, and 11B (3Q)MAS NMR experimentation. Hence, our future investigations will merely target a more direct probing of the medium-range glass structure by utilizing advanced homo/heteronuclear NMR techniques.3,85
Note added after first publication
This article replaces the version published on 25th October 2016, which contained errors in Table 3.Acknowledgements
This work was supported by the Swedish Research Council (contract VR-NT 2014-4667). We thank Baltzar Stevensson for help, and Di Zhang and Renny Mathew for glass synthesis input at an early stage of this project. We also thank R. M. for recording the 29Si NMR spectrum from the S494.0(0) glass.References
- G. Engelhardt and D. Michel, High-Resolution Solid-State NMR Spectroscopy of Silicates and Zeolites, Wiley, Chicester, 1987 Search PubMed.
- G. N. Greaves and S. Sen, Adv. Phys., 2007, 56, 1–166 CrossRef CAS.
- M. Edén, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2012, 108, 177–221 RSC.
- W. Huang, D. E. Day, K. Kittiratanapiboon and M. N. Rahaman, J. Mater. Sci.: Mater. Med., 2006, 17, 583–596 CrossRef CAS PubMed.
- A. Yao, D. Wang, W. Huang, Q. Fu, M. N. Rahaman and D. E. Day, J. Am. Ceram. Soc., 2007, 90, 303–306 CrossRef CAS.
- R. F. Brown, M. N. Rahaman, A. B. Dwilewicz, W. Huang, D. E. Day, Y. Li and B. S. Bal, J. Biomed. Mater. Res., Part A, 2009, 88, 392–400 CrossRef PubMed.
- X. Liu, W. Huang, H. Fu, A. Yao, D. Wang, H. Pan, W. W. Lu, X. Jiang and X. Zhang, J. Mater. Sci.: Mater. Med., 2009, 20, 1237–1243 CrossRef CAS PubMed.
- H. B. Pan, X. L. Zhao, X. Zhang, K. B. Zhang, L. C. Li, Z. Y. Li, W. M. Lam, W. W. Lu, D. P. Wang, W. H. Huang, K. L. Lin and J. Chang, J. R. Soc., Interface, 2010, 7, 1025–1031 CrossRef CAS PubMed.
- M. N. Rahaman, D. E. Day, B. S. Bal, Q. Fu, S. B. Jung, L. F. Bonewald and A. P. Tomsia, Acta Biomater., 2011, 7, 2355–2373 CrossRef CAS PubMed.
- X. Yang, L. Zhang, X. Chen, X. Sun, G. Yang, X. Guo, H. Yang, C. Gao and Z. Gou, J. Non-Cryst. Solids, 2012, 358, 1171–1179 CrossRef CAS.
- Q. Fu, M. N. Rahaman and D. E. Day, J. Am. Ceram. Soc., 2009, 92, 1551–2916 Search PubMed.
- X. Liu, M. N. Rahaman and D. E. Day, J. Mater. Sci.: Mater. Med., 2013, 24, 583–595 CrossRef CAS PubMed.
- L. A. Haro Durand, G. E. Vargas, N. M. Romero, R. Vera-Mesones, J. M. Porto-López, A. R. Boccaccini, M. P. Zago, A. Baldi and A. Gorustovich, J. Mater. Chem. B, 2015, 3, 1142–1148 RSC.
- W. C. Lepry and S. N. Nazhat, Chem. Mater., 2015, 27, 4821–4831 CrossRef CAS.
- L. L. Hench, J. Am. Ceram. Soc., 1991, 74, 1487–1510 CrossRef CAS.
- L. L. Hench and J. M. Polak, Science, 2002, 295, 1014–1017 CrossRef CAS PubMed.
- J. R. Jones, Acta Biomater., 2013, 9, 4457–4486 CrossRef CAS PubMed.
- M. Brink, T. Turunen, R.-P. Happonen and A. Yli-Urpo, J. Mater. Sci.: Mater. Med., 1997, 37, 114–121 CAS.
- B. C. Bunker, G. W. Arnold, D. E. Day and P. J. Bray, J. Non-Cryst. Solids, 1986, 87, 226–253 CrossRef CAS.
- B. C. Bunker and W. H. Casey, The Aqueous Chemistry of Oxides, Oxford University Press, Oxford, 2016 Search PubMed.
- A. Hoppe, N. S. Güldal and A. R. Boccaccini, Biomaterials, 2012, 32, 2757–2774 CrossRef PubMed.
- M. D. O'Donnell, S. J. Watts, R. V. Law and R. G. Hill, J. Non-Cryst. Solids, 2008, 354, 3554–3560 CrossRef.
- M. Edén, J. Non-Cryst. Solids, 2011, 357, 1595–1602 CrossRef.
- H. Gan, P. C. Hess and R. J. Kirkpatrick, Geochim. Cosmochim. Acta, 1994, 58, 4633–4647 CrossRef CAS.
- H. Yamashita, K. Nagata, H. Yoshino, K. Ono and T. Maekawa, J. Non-Cryst. Solids, 1999, 248, 115–126 CrossRef CAS.
- F. Muñoz, L. Montagne, L. Delevoye, A. Durán, L. Pascual, S. Cristol and J.-F. Paul, J. Non-Cryst. Solids, 2006, 352, 28–29 Search PubMed.
- M. Sitarz, K. Bulat and Z. Olejniczak, Vib. Spectrosc., 2012, 61, 72–77 CrossRef CAS.
- R. Dupree, D. Holland, M. G. Mortuza, J. A. Collins and M. W. G. Lockyer, J. Non-Cryst. Solids, 1988, 106, 403–407 CrossRef CAS.
- M. W. G. Lockyer, D. Holland and R. Dupree, Phys. Chem. Glasses, 1995, 36, 22–30 CAS.
- M. W. G. Lockyer, D. Holland and R. Dupree, J. Non-Cryst. Solids, 1995, 188, 207–219 CrossRef CAS.
- V. FitzGerald, D. M. Pickup, D. Greenspan, G. Sarkar, J. J. Fitzgerald, K. M. Wetherall, R. M. Moss, J. R. Jones and R. J. Newport, Adv. Funct. Mater., 2007, 17, 3746–3753 CrossRef CAS.
- R. Mathew, C. Turdean-Ionescu, B. Stevensson, I. Izquierdo-Barba, A. García, D. Arcos, M. Vallet-Regí and M. Edén, Chem. Mater., 2013, 25, 1877–1885 CrossRef CAS.
- R. Mathew, B. Stevensson, A. Tilocca and M. Edén, J. Phys. Chem. B, 2014, 118, 833–844 CrossRef CAS PubMed.
- R. Mathew, B. Stevensson and M. Edén, J. Phys. Chem. B, 2015, 119, 5701–5715 CrossRef CAS PubMed.
- B. Stevensson, R. Mathew and M. Edén, J. Phys. Chem. B, 2014, 118, 8863–8876 CrossRef CAS PubMed.
- A. Tilocca and A. N. Cormack, J. Phys. Chem. B, 2007, 111, 14256–14264 CrossRef CAS PubMed.
- Y. H. Yun and P. J. Bray, J. Non-Cryst. Solids, 1978, 27, 363–380 CrossRef CAS.
- W. J. Dell, P. J. Bray and S. Z. Xiao, J. Non-Cryst. Solids, 1983, 58, 1–16 CrossRef CAS.
- B. C. Bunker, D. R. Tallant, R. J. Kirkpatrick and G. L. Turner, Phys. Chem. Glasses, 1990, 31, 30–41 CAS.
- M. Edén, P. Sundberg and C. Stålhandske, J. Non-Cryst. Solids, 2011, 357, 1587–1594 CrossRef.
- Z. Strnad, Biomaterials, 1992, 13, 317–321 CrossRef CAS PubMed.
- R. Hill, J. Mater. Sci. Lett., 1996, 15, 1122–1125 CrossRef CAS.
- K. L. Geisinger, R. Oestrike, A. Navrotsky, G. L. Turner and R. J. Kirkpatrick, Geochim. Cosmochim. Acta, 1988, 52, 2405–2414 CrossRef CAS.
- A. D. Irwin, J. S. Holmgren and J. Jonas, J. Non-Cryst. Solids, 1988, 101, 249–254 CrossRef CAS.
- S. W. Martin, D. Bain, K. Budhwani and S. Feller, J. Am. Ceram. Soc., 1992, 75, 1117–1122 CrossRef CAS.
- G. El-Damrawi and W. Müller-Warmuth, J. Non-Cryst. Solids, 1992, 146, 137–144 CrossRef CAS.
- L. van Wüllen, W. Müller-Warmuth, D. Papageorgiou and H. J. Pentinghaus, J. Non-Cryst. Solids, 1994, 171, 53–67 CrossRef.
- S. Wang and J. F. Stebbins, J. Am. Ceram. Soc., 1999, 82, 1519–1528 CrossRef CAS.
- R. Martens and W. Müller-Warmuth, J. Non-Cryst. Solids, 2000, 265, 167–175 CrossRef CAS.
- L.-S. Du and J. F. Stebbins, J. Non-Cryst. Solids, 2003, 315, 239–255 CrossRef CAS.
- L.-S. Du and J. F. Stebbins, J. Phys. Chem. B, 2003, 107, 10063–10076 CrossRef CAS.
- L.-S. Du and J. F. Stebbins, Chem. Mater., 2003, 15, 3913–3921 CrossRef CAS.
- M. M. Smedskjaer, J. C. Mauro, R. E. Youngman, C. L. Hogue, M. Potuzak and Y. Yue, J. Phys. Chem. B, 2011, 115, 12930–12946 CrossRef CAS PubMed.
- D. Möncke, G. Tricot, A. Winterstein-Beckmann, L. Wondraczek and E. I. Kamitsos, Phys. Chem. Glasses: Eur. J. Glass Sci. Technol., Part B, 2015, 56, 203–211 CrossRef.
- R. E. Youngman and J. W. Zwanziger, J. Non-Cryst. Solids, 1994, 168, 293–297 CrossRef CAS.
- F. Angeli, T. Charpentier, D. De Ligny and C. Cailleteau, J. Am. Ceram. Soc., 2010, 93, 2693–2704 CrossRef CAS.
- L. Frydman and J. S. Harwood, J. Am. Chem. Soc., 1995, 117, 5367–5368 CrossRef CAS.
- J.-P. Amoureux, C. Fernandez and S. Steurnagel, J. Magn. Reson., Ser. A, 1996, 123, 116–118 CrossRef CAS PubMed.
- T. Vosegaard, P. Florian, P. J. Grandinetti and D. Massiot, J. Magn. Reson., 2000, 143, 217–222 CrossRef CAS PubMed.
- P. K. Madhu, A. Goldbourt, L. Frydman and S. Vega, Chem. Phys. Lett., 1999, 307, 41–47 CrossRef CAS.
- D. J. States, R. A. Haberkorn and D. J. Ruben, J. Magn. Reson., 1982, 48, 286–292 CAS.
- Y. Millot and P. P. Man, Solid State Nucl. Magn. Reson., 2002, 21, 21–43 CrossRef CAS PubMed.
- H. Grussaute, L. Montagne, G. Palavit and J. L. Bernard, J. Non-Cryst. Solids, 2000, 263, 312–317 CrossRef.
- E. Leonova, I. Izquierdo-Barba, D. Arcos, A. López-Noriega, N. Hedin, M. Vallet-Regí and M. Edén, J. Phys. Chem. C, 2008, 112, 5552–5562 CAS.
- F. Fayon, C. Duée, T. Poumeyrol, M. Allix and D. Massiot, J. Phys. Chem. C, 2013, 117, 2283–2288 CAS.
- A.-R. Grimmer, M. Mägi, M. Hähnert, H. Stade, A. Samoson, W. Weiker and E. Lippmaa, Phys. Chem. Glasses, 1984, 25, 105–109 CAS.
- H. Maekawa, T. Maekawa, K. Kawamura and T. Yokokawa, J. Non-Cryst. Solids, 1991, 127, 53–64 CrossRef CAS.
- D. Massiot, C. Bessada, J. P. Coutures and F. Taulelle, J. Magn. Reson., 1990, 90, 231–242 CAS.
- L. van Wüllen and W. Müller-Warmuth, Solid State Nucl. Magn. Reson., 1993, 2, 279–284 CrossRef.
- P. R. Bodart, J. Magn. Reson., 1998, 133, 207–209 CrossRef CAS PubMed.
- S. Wegner, L. van Wüllen and G. Tricot, Solid State Sci., 2010, 12, 428–439 CrossRef CAS.
- J. F. Stebbins, P. Zhao and S. Kroeker, Solid State Nucl. Magn. Reson., 2000, 16, 9–19 CrossRef CAS PubMed.
- S. Kroeker and J. F. Stebbins, Inorg. Chem., 2001, 40, 6239–6246 CrossRef CAS PubMed.
- P. M. Aguiar and S. Kroeker, Solid State Nucl. Magn. Reson., 2005, 27, 10–15 CrossRef CAS PubMed.
- P. M. Aguiar and S. Kroeker, J. Non-Cryst. Solids, 2007, 353, 1834–1839 CrossRef CAS.
- M. R. Hansen, T. Vosegaard, H. J. Jakobsen and J. Skibsted, J. Phys. Chem. A, 2004, 108, 586–594 CrossRef CAS.
- J. Banerjee, G. Ongie, J. Harder, T. Edwards, C. Larson, S. Sutton, A. Moeller, A. Basu, M. Affatigato, S. Feller, M. Kodama, P. M. Aguiar and S. Kroeker, J. Non-Cryst. Solids, 2006, 352, 674–678 CrossRef CAS.
- D. Holland, S. A. Feller, T. F. Kemp, M. E. Smith, A. P. Howes, D. Winslow and M. Kodama, Phys. Chem. Glasses: Eur. J. Glass Sci. Technol., Part B, 2007, 48, 1–8 CAS.
- O. L. G. Alderman, D. Iuga, A. P. Howes, K. J. Pike, D. Holland and R. Dupree, Phys. Chem. Chem. Phys., 2013, 15, 8208–8221 RSC.
- J. C. Phillips and R. Kerner, J. Chem. Phys., 2008, 128, 174506 CrossRef CAS PubMed.
- A. P. Howes, N. M. Vedishcheva, A. Samoson, J. V. Hanna, M. E. Smith, D. Holland and R. Dupree, Phys. Chem. Chem. Phys., 2011, 13, 11919–11928 RSC.
- M. M. Smedskjaer, R. E. Youngman and J. C. Mauro, Appl. Phys. A, 2014, 116, 491–504 CrossRef CAS.
- T. Kokubo, H. Kushitani, S. Sakka, T. Kitsugi and T. Yamamuro, J. Biomed. Mater. Res., 1990, 24, 721–734 CrossRef CAS PubMed.
- I. Lebecq, F. Désanglois, A. Leriche and C. Follet-Houttemane, J. Biomed. Mater. Res., 2007, 83, 156–168 CrossRef CAS PubMed.
- M. Edén, Solid State Nucl. Magn. Reson., 2009, 36, 1–10 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2016 |