
Theoretical study and design of cyclometalated platinum complexes bearing innovatively a highly-rigid terdentate ligand with carboranyl as a chelating unit†
Abstract
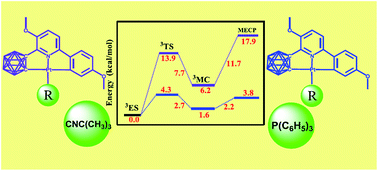
Density functional theory (DFT) and time-dependent density functional theory (TD-DFT) were employed to explore the electronic structures and phosphorescence properties of synthesized terdentate Pt(II) complexes bearing highly-rigid 3,6-bis(p-anizolyl)-2-carboranyl-pyridine as a cyclometalated ligand and triphenylphosphine (1) or t-butylisonitrile (2) as ancillary ligand. To understand the marked difference in phosphorescence quantum efficiency between 1 and 2, the relaxation dynamics of excited states were elucidated in detail. Aiming to formulate the radiative relaxation, the zero-field splitting (ZFS) and the radiative decay rate constant (kr) were calculated by SOC-perturbed TDDFT (pSOC-TDDFT). Meanwhile, the temperature-independent non-radiative relaxation was analyzed by calculating the Huang–Rhys factor (S), the SOC interaction between the emitting state and the ground state. While the temperature-dependent non-radiative decay mechanism was studied by depicting the thermal deactivation process via a metal-centered excited 3MC state. Based on the results, 1 and 2 show a few differences in their temperature-independent non-radiative rates. However, the activation barrier for the population of non-emissive 3MC is greatly raised for complex 2. Therefore, the temperature-dependent non-radiative decay behavior of 2 is considerably suppressed, which ultimately leads to a substantially enhanced phosphorescence quantum efficiency for 2. To further tune the emission wavelength towards blue, four new complexes 3–6 were theoretically designed by modifying the terdentate ligand with azole groups based on the parent complex 2. As a result, pyrazole modified complex 4 stands out with enhanced deep-blue phosphorescence located at 434 nm.
 Please wait while we load your content...
Please wait while we load your content...

