DOI:
10.1039/C6RA14257E
(Paper)
RSC Adv., 2016,
6, 68675-68684
Highly water-dispersible magnetite-supported Pd nanoparticles and single atoms as excellent catalysts for Suzuki and hydrogenation reactions†
Received
1st June 2016
, Accepted 3rd July 2016
First published on 4th July 2016
Abstract
The molecule 4-(diphenylphosphino)benzoic acid (dpa) anchored on the surface of magnetite nanoparticles permits the easy capture of palladium ions that are deposited on the surface of the magnetite nanoparticles after reduction with NaBH4. Unexpectedly, a significant fraction of dpa is removed in this process. Samples of Fe3O4dpa@Pdx containing different Pd loadings (x = 0.1, 0.3, 0.5 and 1.0 wt%) were prepared, and their catalytic efficiency for the Suzuki C–C coupling reaction was studied. The best catalyst was Fe3O4dpa@Pd0.5, which gave the highest TOF published to date for the reaction of bromobenzene with phenylboronic acid in a mixture of ethanol/water (1/1). Interestingly, the same reaction carried out in water also produced excellent yields of the resulting C–C coupling product. The behaviour of other bromide aryl molecules was also investigated. The best catalytic results for the aqueous phase reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) were obtained using Fe3O4dpa@Pd0.1. The presence of Pd SACs (single atom catalysts) seems to be responsible for this performance. In contrast, the same Fe3O4dpa@Pd0.1 catalyst is absolutely inactive for the hydrogenation of styrene in ethanol.
Introduction
For the last few decades, Pd nanoparticles (NPs) have attracted enormous attention as transition metal catalysts in an array of transformation processes in organic chemistry, mainly for C–C cross-coupling reactions.1 Given that easy aggregation of nanoparticles can affect their catalytic behaviour, it is crucial to protect them with appropriate linkers, namely, thiol-protected linkers,2,3 dendrimers,4 or dendrons,5 or by the use of ionic liquids.6 Another strategy involves the immobilization of the Pd NPs on solid supports, usually SiO2, TiO2 and C, prepared by different procedures and having different areas and morphologies.7 This interest has increased in recent years with the easy deposition of Pd onto the surface of magnetite NPs, resulting in magnetically separable catalytic systems that avoid the requirement of catalyst filtration after completion of the reaction. In addition, the nanocatalyst can be efficiently recycled and reused in the vast majority of examples.8 For anchoring small and homogeneous nanoparticles, it is worthwhile to previously functionalize the magnetite surface with linkers, generally terminated with amino9,10 or phosphine units.11 However, the benefits produced by the linkers on the deposition of the metal catalysts should be contrasted with the potential catalytic inhibition produced by their steric hindrance, which obstructs the approach of the reagents to the catalyst. In this context, it is evident that the possibility of partial removal of the linker after metal immobilization becomes a desirable condition.
Very recently, we have reported the synthesis and use of 4-mercaptophenyldiphenylphosphine (Sdp) as a linker to immobilize Pd nanoparticles onto the surface of magnetite nanoparticles.12 Subsequent treatment of the nanoparticles with an aqueous hydrogen peroxide solution permitted the removal of 70 wt% of Sdp. The partial loss of the linker made the resulting nanoparticles catalytically more efficient for the Suzuki–Miyaura C–C cross coupling reaction and for hydrogenation of 4-nitrophenol and styrene.
In this paper, we expand this study to the formation of Pd/magnetite nanoparticles and single atoms using partially water soluble and commercially available 4-(diphenylphosphino)benzoic acid (Fig. 1) as a linker in order to make the above processes “greener” by minimizing the use of (toxic) organic solvents. In addition to this, the increased demand for scarce metals such as palladium and their future availability13 is a matter of concern that may be dispelled if control of the palladium particle size is achieved for proper atomic economy.
 |
| | Fig. 1 4-(Diphenylphosphino)benzoic acid (dpa). | |
Results and discussion
Synthesis of the catalysts
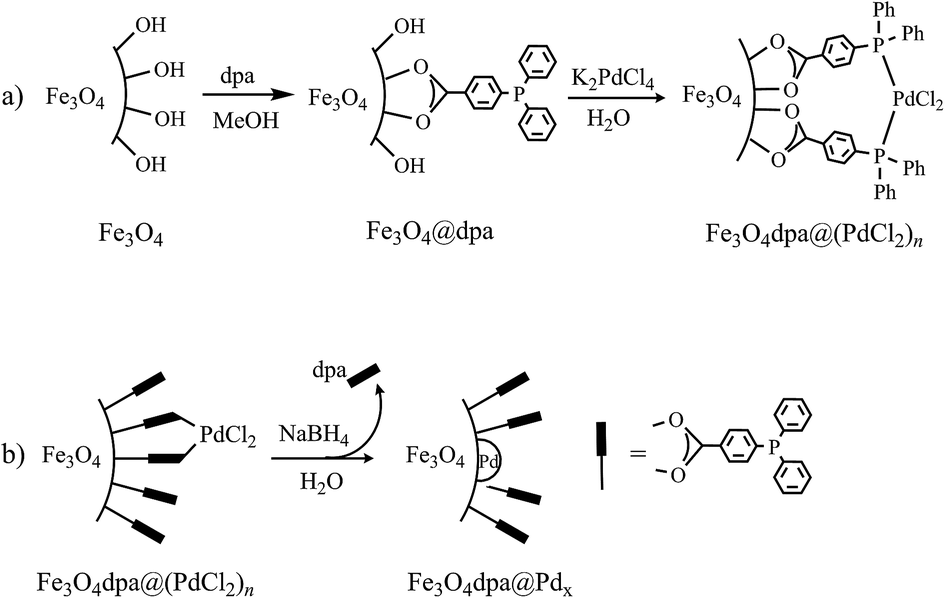
The starting bare magnetite nanoparticles were synthesized following a literature procedure.14 Functionalized magnetite nanoparticles, Fe3O4dpa, were obtained by sonication of Fe3O4 in methanol in the presence of dpa for 2 hours (Scheme 1a). Elemental analyses (C, H) of the resulting nanoparticles showed that the load of the organic component was about 3.2 wt%. The next step involved immobilizing Pd onto the magnetite surface (Scheme 1b). Thus, Fe3O4dpa was dispersed in water and sonicated for 10 min. Then, a water solution of K2[PdCl4] followed by another of sodium borohydride were added to obtain the Fe3O4dpa@Pdx nanoparticles, where x is the weight percentage of Pd in the NPs. With this method, we prepared several samples with different Pd contents (0.1%, 0.3%, 0.5% and 1.0%). The first one was included in order to obtain magnetite NPs decorated exclusively with isolated palladium single atoms. These species are denoted as SACs (single-atom catalysts)15 and exhibit generally exceptional catalytic activity.
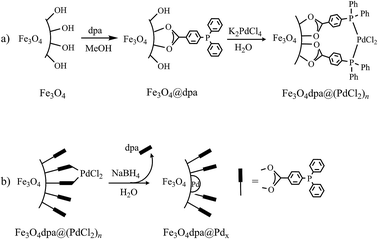 |
| | Scheme 1 Synthesis of catalysts: (a) MNPs functionalization with dpa and Pd(II) capture and (b) deposition of palladium nanoparticles and partial loss of dpa. | |
A crucial and surprising observation in the course of the fabrication of Fe3O4dpa@Pdx NPs was the significant fraction of dpa lost during the reduction process with NaBH4. For example, Fe3O4dpa@Pd0.5 only retained 1.7% dpa from 3.2% of the starting Fe3O4dpa NPs. That is, dpa favours the dispersion of Pd NPs on the magnetite surface, and then the ligand undergoes partial elimination without the use of other agents. Consequently, the low linker content in Fe3O4dpa@Pdx NPs is expected to yield excellent catalytic activity, which we have explored in the Suzuki–Miyaura reaction, the reduction of 4-nitrophenol and the styrene hydrogenation processes.
Characterization of Fe3O4dpa@Pdx NPs
The FT-IR spectra of the catalysts are practically identical, showing a sharp peak at 594 cm−1, which is the IR signature for ferrite particles, and weak signals at 1100 cm−1 and 2921 cm−1 due to aryl C–H vibrations. The Pd content in Fe3O4dpa@Pdx was determined by ICPoes (Inductive Coupled Plasma optical emission spectroscopy) measurements, and the dpa content was estimated by OEA (Organic Elemental Analysis). The analytical composition of the hybrids, as well as of the NPs loaded with Pd in the absence of linker, is shown in Table 1.
Table 1 Composition of magnetite–dpa–Pd hybrids
| Catalyst |
% dpa (nmol mg−1 NPs) |
% Pd |
| Fe3O4dpa |
3.2 (105) |
0 |
| Fe3O4@Pd |
0 (0) |
0.68 |
| Fe3O4dpa@Pd0.1 |
2.4 (78.7) |
0.097 |
| Fe3O4dpa@Pd0.3 |
1.9 (63.0) |
0.32 |
| Fe3O4dpa@Pd0.5 |
1.7 (55.2) |
0.51 |
| Fe3O4dpa@Pd1.0 |
1.1 (35.0) |
1.01 |
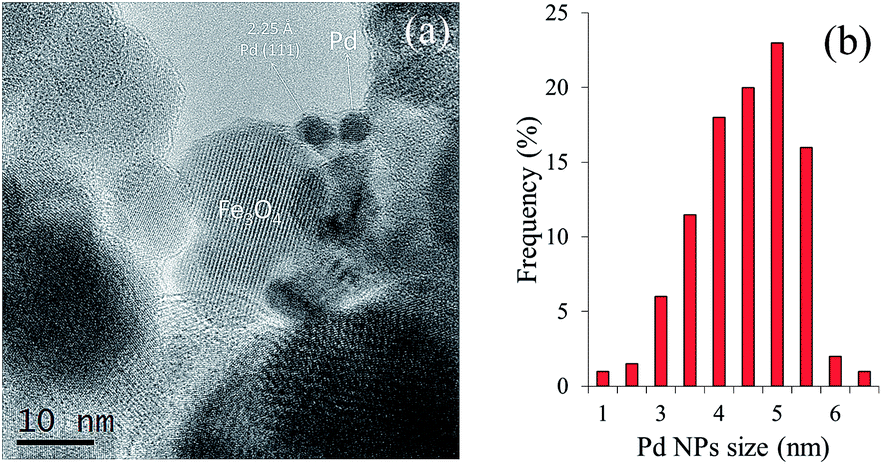
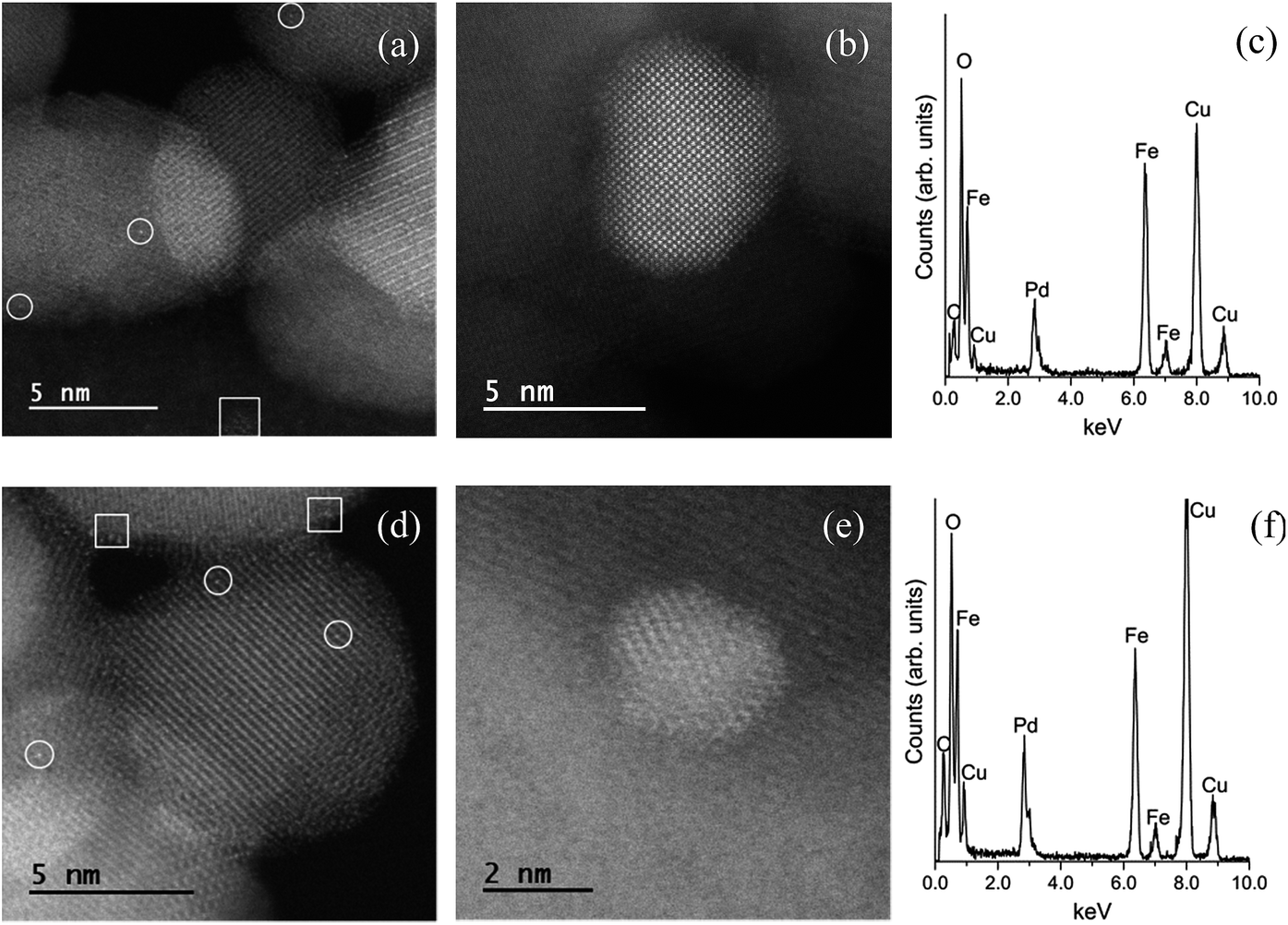
Images of the hybrid Fe3O4dpa@Pdx nanoparticles were obtained by TEM (for x = 1.0) and HAADF-STEM (High-Angle Annular Dark-Field Scanning Transmission Electron Microscopy) (for x = 0.1, 0.3, and 0.5). The samples display spherical Fe3O4 nanoparticles with an average particle size of around 12 nm. In addition, smaller Pd species appear to be immobilized on the magnetite surface. As expected, the total Pd load determines the Pd nanoparticle size. Thus, for x = 1.0, the images clearly reveal the presence of nanoparticles with an average size of 4.5 nm (Fig. 2). In contrast, when the Pd loading is 0.1 wt%, all Pd species exist exclusively as isolated single atoms; neither subnanometre clusters nor nanoparticles were detected (Fig. 3). Samples containing intermediate palladium contents, x = 0.3 and 0.5, evidenced numerous single metal atoms accompanied by other subnanometer clusters and larger nanoparticles. This fact is somewhat surprising because the presence of large nanoparticles of ∼5 nm along with a significant number of Pd SACs is unprecedented (Fig. 4). The presence of palladium was confirmed by energy-dispersive X-ray spectroscopy (EDS) measurements; sharp Pd Lα1 and Lβ1 peaks are clearly visible at 2.84 and 2.99 keV, respectively (Fig. 4c and f).
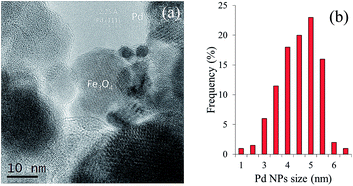 |
| | Fig. 2 (a) TEM images of Fe3O4dpa@Pd1.0. (b) Histogram of the Pd particle size distribution. | |
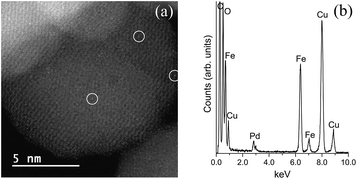 |
| | Fig. 3 (a) HAADF-STEM images of Fe3O4dpa@Pd0.1. Isolated Pd atoms (white circles) are uniformly dispersed on the magnetite support. (b) EDS spectrum. The Cu and C signals originate from the TEM grid. | |
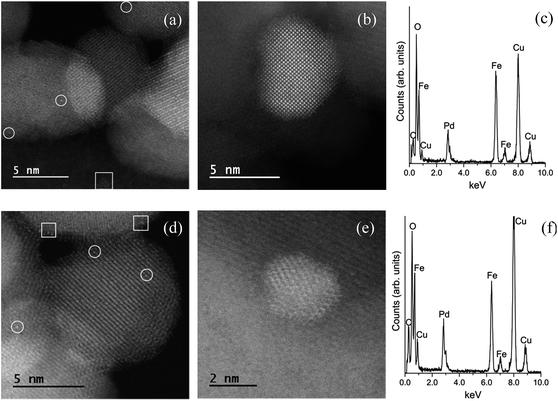 |
| | Fig. 4 (a and b) HAADF-STEM images of Fe3O4dpa@Pd0.3 showing the presence of different Pd species. (c) EDS spectrum of Fe3O4dpa@Pd0.3. (d and e) HAADF-STEM images of Fe3O4dpa@Pd0.5 showing the presence of different Pd species. (f) EDS spectrum of Fe3O4dpa@Pd0.5. The circles and squares in the images indicate Pd single atoms and clusters/nanoparticles, respectively. | |
X-ray Photoelectron Spectroscopy (XPS) was used to determine the surface elemental composition of the Fe3O4dpa@Pdx nanocomposites (Fig. S1†). The surface Pd/Fe atomic ratios are compiled in Table 2. For the Fe3O4dpa@Pdx catalysts with x = 0.1, 0.3 and 0.5, the Pd/Fe ratio progressively increases from Pd/Fe = 0.003 to 0.010, as expected, since all the Pd single atoms and subnanometer clusters observed by HAADF-STEM are sampled. However, for the Fe3O4dpa@Pd1.0 sample, the Pd/Fe ratio is lower because not all the Pd in the Pd nanoparticles is sampled due to the larger dimensions of the Pd nanoparticles, in total accordance with the high-resolution TEM results discussed above. The oxidation state of Pd in each catalyst is also included in Table 2, where the relative contributions from Pd(0), Pd(II) and Pd(IV) species and their binding energies (Pd 3d5/2) are indicated. There is a clear trend between the oxidation state of Pd and its content in the catalysts; the higher the Pd loading, the more oxidized the Pd. The sample with the highest Pd content, Fe3O4dpa@Pd1.0, exhibits photoemission peaks ascribed to Pd(IV), which confirms the tendency of Pd oxidation in this series. On the other hand, the sample Fe3O4dpa@Pd0.1, which only contains Pd single atoms, exhibits a Pd(0) component at a significantly higher binding energy value (335.8 vs. 335.2 to 335.3 eV), which suggests that there is an electronic transfer from the Pd single atoms to the Fe3O4 support, in accordance with previous reports.16,17
Table 2 Pd/Fe surface atomic ratios, percentage of Pd species and binding energies obtained by XPS
| Catalyst |
Pd/Fe |
% (BE Pd 3d5/2, eV) |
| Pd(0) |
Pd(II) |
Pd(IV) |
| Fe3O4@Pd |
0.006 |
42 (335.2) |
58 (337.5) |
— |
| Fe3O4dpa@Pd0.1 |
0.003 |
48 (335.8) |
52 (337.3) |
— |
| Fe3O4dpa@Pd0.3 |
0.005 |
30 (335.3) |
70 (337.4) |
— |
| Fe3O4dpa@Pd0.5 |
0.010 |
18 (335.2) |
82 (337.4) |
— |
| Fe3O4dpa@Pd1.0 |
0.007 |
18 (335.3) |
58 (337.5) |
24 (338.7) |
Catalytic studies
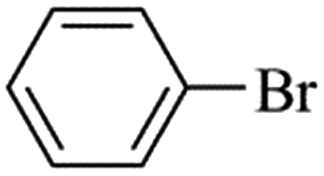
Suzuki–Miyaura reaction.
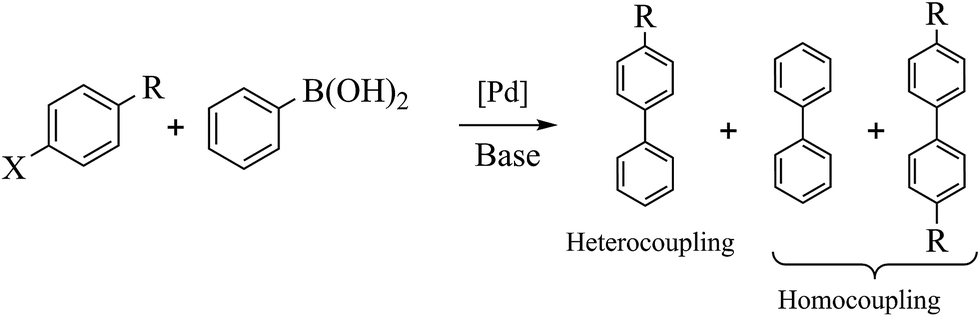
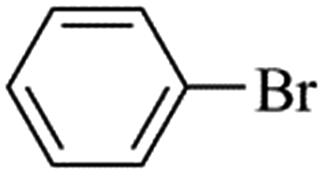
Palladium-catalyzed Suzuki–Miyaura cross-coupling is a well-known process for the synthesis of biaryls using aryl halides with arylboronic (Scheme 2) acids; it has many applications in agrochemicals, natural products and pharmaceutical intermediates.18 To test the catalytic activity of Fe3O4dpa@Pdx NPs, the reaction between bromobenzene and phenylboronic acid (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was analysed and optimized in terms of time, temperature (Table S1†), base (Table S2†) and solvent (Table S3†). The best results were obtained with Fe3O4dpa@Pd0.5 NPs after 20 min at 65 °C in EtOH/water (1:1) and K3PO4. Table 3 lists the catalytic results. Note that Fe3O4dpa NPs in the absence of Pd (entry 1) showed catalytic inactivity, while entry 2 revealed the poor catalytic behaviour of the NPs loaded with Pd in the absence of linker. Entries 3 to 6 display the results obtained with the remaining Pd-containing NPs. Clearly, the best catalyst is the sample containing 0.5% Pd (entry 5). The catalyst consisting only of single atoms (Fe3O4dpa@Pd0.1) (entry 3) and Fe3O4dpa@Pd0.3 (entry 4) are also excellent, although the sample loaded with 1.0 Pd% is clearly less efficient (entry 6) due the large nanoparticle size.
:1) was analysed and optimized in terms of time, temperature (Table S1†), base (Table S2†) and solvent (Table S3†). The best results were obtained with Fe3O4dpa@Pd0.5 NPs after 20 min at 65 °C in EtOH/water (1:1) and K3PO4. Table 3 lists the catalytic results. Note that Fe3O4dpa NPs in the absence of Pd (entry 1) showed catalytic inactivity, while entry 2 revealed the poor catalytic behaviour of the NPs loaded with Pd in the absence of linker. Entries 3 to 6 display the results obtained with the remaining Pd-containing NPs. Clearly, the best catalyst is the sample containing 0.5% Pd (entry 5). The catalyst consisting only of single atoms (Fe3O4dpa@Pd0.1) (entry 3) and Fe3O4dpa@Pd0.3 (entry 4) are also excellent, although the sample loaded with 1.0 Pd% is clearly less efficient (entry 6) due the large nanoparticle size.
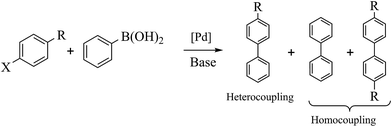 |
| | Scheme 2 Suzuki–Miyaura coupling. | |
Table 3 Activity of different catalysts in Suzuki–Miyaura couplinga
| Entry |
Catalyst |
TOF (h−1) |
|
TOF (mol product/mol catalyst·time). Bromobenzene (3 mmol), phenylboronic acid (3.6 mmol), K3PO4 (9 mmol), catalyst (9.4 × 10−5 mmol Pd), 65 °C, EtOH:water.
|
| 1 |
Fe3O4dpa |
0 |
| 2 |
Fe3O4@Pd |
870 |
| 3 |
Fe3O4dpa@Pd0.1 |
51200 |
| 4 |
Fe3O4dpa@Pd0.3 |
64600 |
| 5 |
Fe3O4dpa@Pd0.5 |
110000 |
| 6 |
Fe3O4dpa@Pd1.0 |
1630 |
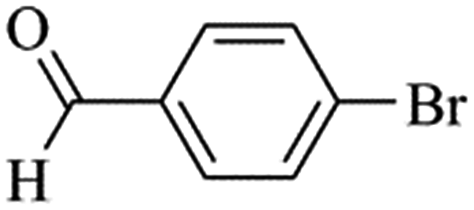
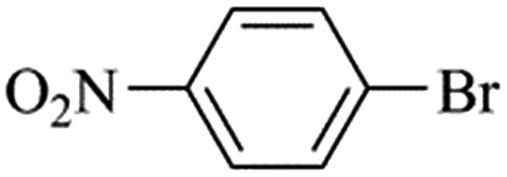
We next proceeded to examine the scope and limitation of the catalysts. Using Fe3O4dpa@Pd0.5 NPs, several bromo-aryl substrates were reacted with phenylboronic acid (1:1), and the coupling products were obtained in excellent yields (Table 4). Note that the Pd used is as low as 9.4 × 10−5 eq. To our knowledge, the TOFs exhibited with Fe3O4 dpa@Pd0.5 NPs for the Suzuki–Miyaura C–C coupling are by far the highest reported to date.
Table 4 Substrate effect on Suzuki–Miyaura coupling with Fe3O4dpa@Pd0.5 as catalysta
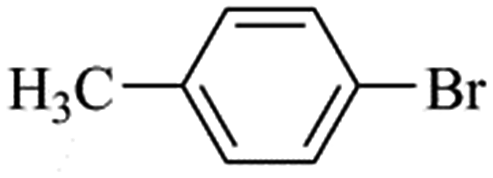
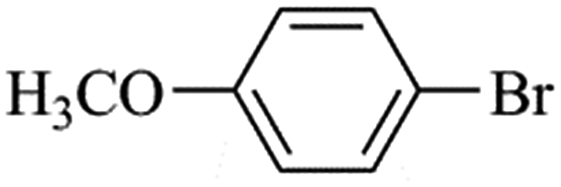
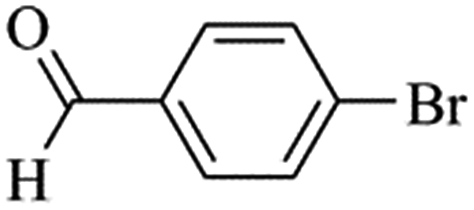
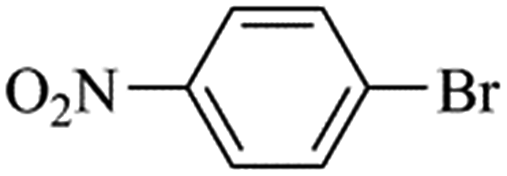
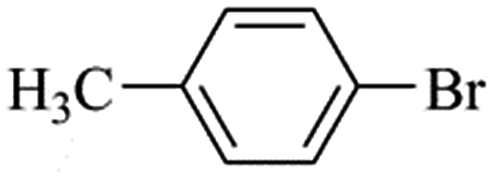
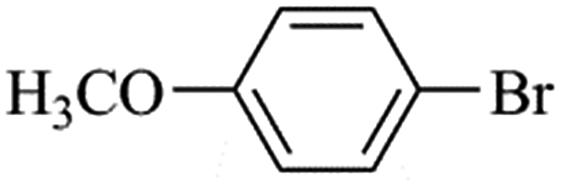
| Bromoaryl |
TOF (h−1) |
|
Bromoaryl (3 mmol), phenylboronic acid (3.6 mmol), K3PO4 (9 mmol), catalyst (9.4 × 10−5 mmol Pd), 65 °C, EtOH:water (60 mL).
|
|
110000 |
|
96300 |
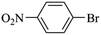
|
35000 |
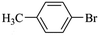
|
38400 |
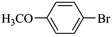
|
28300 |
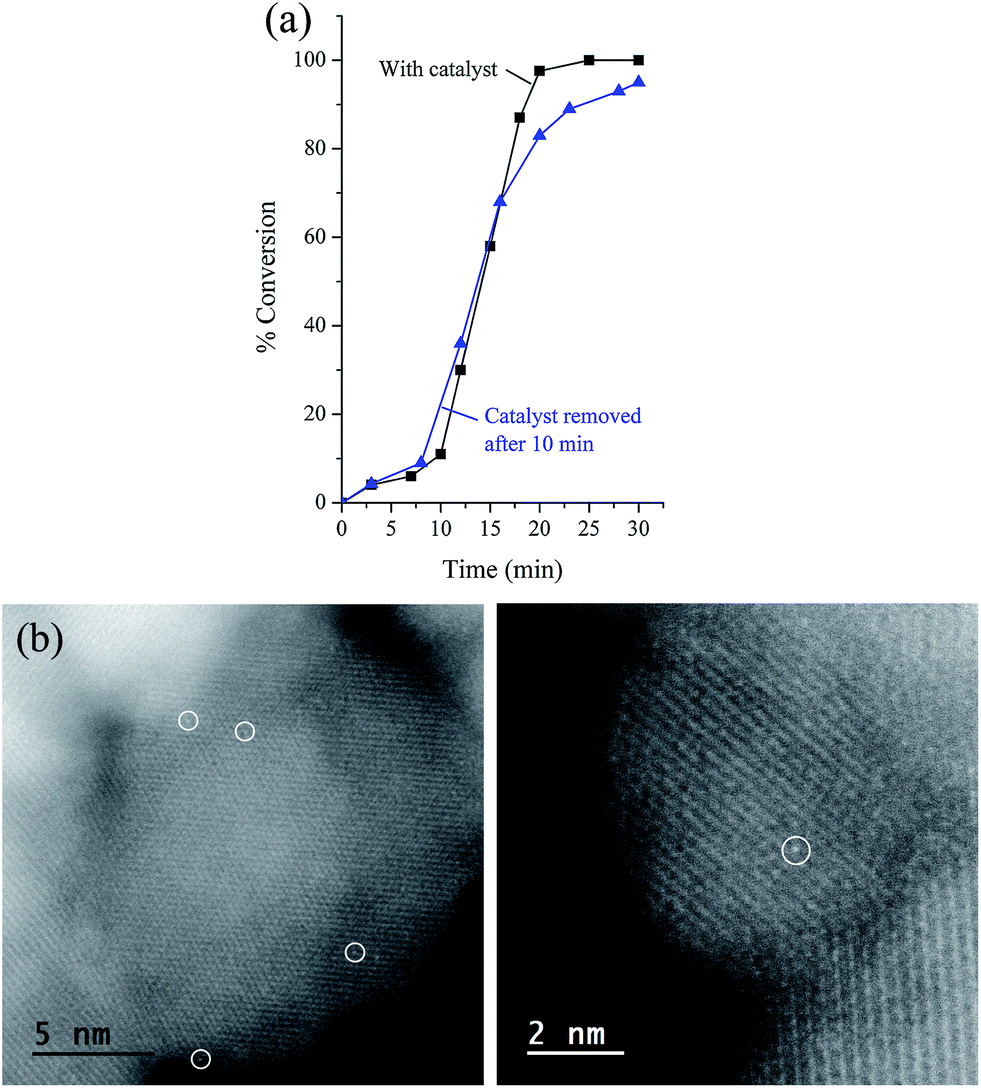
The lifetime of catalytic systems and their reusability is important for practical applications. The reusability of the catalyst was examined following the reaction of bromobenzene and phenylboronic acid. In detail, the NPs were collected after completion of the first reaction using an external magnet and were washed with ethanol and dried. Then, the recovered catalyst nanoparticles were used for the next round by mixing them with a new substrate, base, and solvent. The third round was carried out following the same procedure, and the results are displayed in Table 5. As can be seen, the loss of the catalytic efficiency is remarkably high and greater than that detected, for example, by employing Sdp linker.12 The strong decrease of the catalytic activity is concomitant with the reduction of Pd concentration (33 wt%) found in the residual NPs. The measured leaching of Pd suggests that the catalytic process is essentially homogeneous, which was subsequently confirmed by a hot filtration test. Thus, bromobenzene and phenylboronic acid were reacted at 65 °C. After 10 min, the solid catalyst was separated magnetically and the filtrate was transferred to another Schlenk flask. Without the presence of catalyst, the reaction progressed to 95% conversion after 30 min. This result is in good accord with recently reported data arguing that palladium species leached out from nanoparticles into the reaction mixture are indeed the actual catalytic species instead of the original Pd NPs.19 In addition, the HAADF-STEM images of the catalyst after 3 cycles showed the presence of only palladium single atoms (Fig. 5), strengthening the idea that strong bonding between Pd single atoms and naked Fe3O4 takes place.16 This behavior explains the retained activity of the catalyst after several cycles.
Table 5 Reuse of Fe3O4dpa@Pd0.5 as catalyst in the Suzuki–Miyaura couplinga
| Cycle |
TOF (h−1) |
|
Bromobenzene (3 mmol), phenylboronic acid (3.6 mmol), K3PO4 (9 mmol), catalyst (9.4 × 10−5 mmol Pd), 65 °C, EtOH:water 1:1.
|
| 1 |
110000 |
| 2 |
19200 |
| 3 |
4800 |
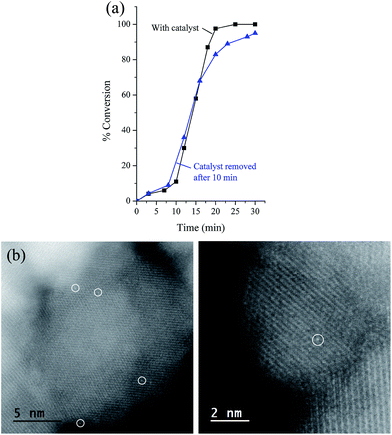 |
| | Fig. 5 (a) Conversion of bromobenzene after Fe3O4dpa@Pd0.5 removal. (b) HAADF-STEM of Fe3O4dpa@Pd0.5 after the third recycling process. Only Pd single atoms were present (white circles). | |
The Suzuki reaction is difficult to achieve with chloroaryl substrates. In this paper, only traces of the final product were observed by reaction of chlorobenzene and 4-chlorotoluene with phenylboronic acid in the presence of Fe3O4dpa@Pd0.5.
Catalysis in neat water.
There are very few articles on the application of magnetically separable Pd-catalyzed Suzuki–Miyaura reaction in neat water; in some of the examples, the process required the use of large amounts of ionic liquids or phase transfer reagents.20 That is, there is still much room for developing new magnetically recoverable catalysts that can be used in pure water. Here, we report the catalytic behaviour of Fe3O4dpa@Pd0.5 NPs in water for the Suzuki coupling under air atmosphere. Although the boronic acids are soluble in water, the efficiency of the Suzuki reaction in neat water can be affected by side reactions, mainly, homocoupling reactions of the boronic acids. The reaction of bromobenzene and phenylboronic acid was used as a model reaction and was optimized in terms of time and temperature. We next examined the scope of this process using several substrates (Table 6). It is well known that the coupling reactions of aryl bromides containing electron-donating groups proceeds more efficiently than that of those having electron-withdrawing groups. This trend is not observed in our results, because the solubility of the substrates is the predominant factor in water. The formation of homocoupling products was negligible. The overall results shown in Table 7 are exceptional in terms of TOFs and selectivity, surpassing all those reported earlier; however, the reusability is very limited. For example, using bromobenzaldehyde as a reagent, it was found that after the third cycle the TOF decreased from 5030 to 106 h−1. Palladium leaching was clearly evidenced by a hot filtration test in which the important loss (28 wt%) of Pd content (by ICPoes analysis) in residual NPs confirmed the homogeneous nature of the catalytic process.
Table 6 Suzuki–Miyaura coupling with different substrates using Fe3O4dpa@Pd0.5 as the catalysta
| Bromoaryl |
TOF (h−1) |
|
Bromoaryl (0.5 mmol), phenylboronic acid (0.6 mmol), K3PO4 (1.5 mmol), 2.0 × 10−4 mmol Pd, 65 °C, water, air atmosphere.
|
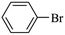
|
3150 |
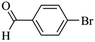
|
5030 |
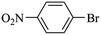
|
0 |
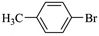
|
1479 |
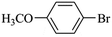
|
475 |
Table 7 Comparison of Suzuki–Miyaura couplings catalyzed by Pd NPs from the literaturea
| Support |
Substrate |
Solvent |
Base |
Temperature (°C) |
TOF (h−1) |
Ref. |
|
4-BN: 4-bromonitrobenzene. 4-BA: 4-bromoanisol. BB, bromobenzene.
|
| Fe3O4dpa@Pd0.5 |
BB |
EtOH:H2O |
K3PO4 |
65 |
110000 |
This work |
| Fe3O4dpa@Pd0.5 |
BB |
H2O |
K3PO4 |
65 |
1470 |
This work |
| Fe3O4Sdp@Pd0.5 |
4-BA |
EtOH:H2O |
KOH |
65 |
60 |
12
|
| γ-Fe2O3/dendrimer |
4-BN |
THF/Triton |
NaOH |
65 |
41.7 |
21
|
| Fe3O4–SiO2/phosphine |
4-BN |
H2O/DMF |
K2CO3 |
25 |
13.1 |
22
|
| Fe3O4–SiO2/phosphine–Pd(II) |
4-BA |
MeOH |
K3PO4 |
60 |
132 |
23
|
| Graphitic carbon nitride/none |
4-BN |
EtOH:H2O |
KOH |
25 |
232 |
24
|
| No support/ionic phosphine |
4-BA |
Glycerol |
t-BuOK |
100 |
50 |
25
|
| Fe3O4–SiO2/iminophosphine |
4-BA |
Toluene |
KOH |
100 |
100 |
11b
|
| Fe3O4/dopamine–phosphine |
4-BA |
Toluene |
KOH |
100 |
33 |
11a
|
| NiFe2O4/dopamine |
4-BA |
DMF |
K3PO4 |
65 |
0.4 |
26
|
| Carbon |
4-BA |
EtOH:H2O |
NaOH |
65 |
171 |
24
|
| Phosphine dendrimer |
4-BD |
Dioxane |
K3PO4 |
100 |
82.5 |
27
|
| Fe3O4 |
4-BA |
Toluene |
Na2CO3 |
130 |
0.01 |
28
|
| Click dendrimer |
BB |
EtOH:H2O |
K3PO4 |
28 |
16500 |
29
|
| Fe3O4–SiO2/TEG-imidazolium |
BB |
H2O |
K2CO3 |
60 |
437 |
30
|
| Multi-walled carbon nanotubes/Pd/PdO |
BB |
H2O |
i-Pr2NH |
100 |
250 |
31
|
| Fe3O4/triazole-acid |
BB |
EtOH:H2O |
K2CO3 |
70 |
2920 |
32
|
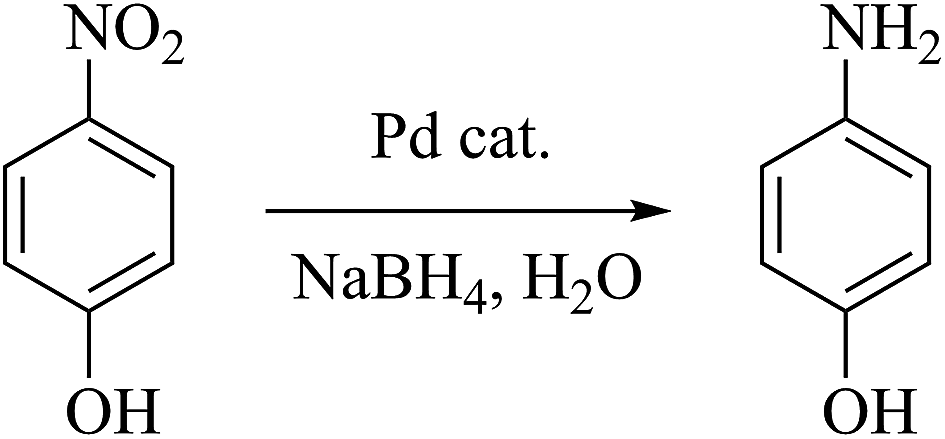
Hydrogenation of 4-nitrophenol.
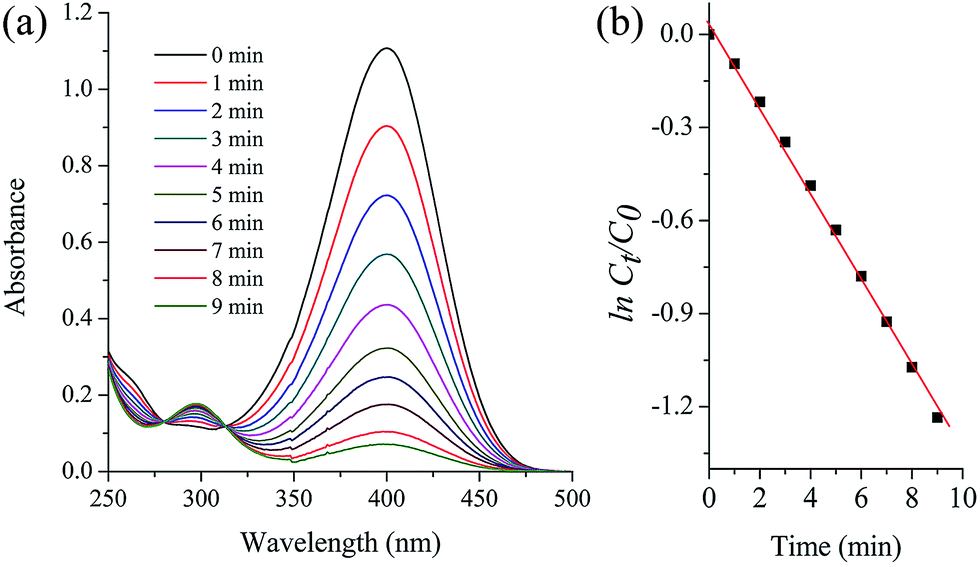
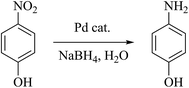
Nitrophenol is a by-product produced from pesticides and synthetic dyes that causes damage to the human central nervous system; thus, its removal from the environment is a crucial task.33 Recently, FeOx-supported platinum SAC34 and carbon nitride–palladium SAC35 have been reported to show excellent activity for the hydrogenation of nitroarenes; however, to our knowledge, Pd SACs supported on magnetite NPs have not been explored for the reduction of 4-nitrophenol in water. The aqueous phase reduction reaction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) (Scheme 3) using metal-supported NPs is a model reaction that permits a comparison of the efficiency of the employed catalysts. Esumi et al. have proposed that the catalytic reduction of 4-NP proceeds in two steps: (i) diffusion and adsorption of 4-NP to the catalyst surface and (ii) electron transfer mediated by the catalyst surface from BH4− to 4-NP.36 The rapid reduction generally is carried out using NaBH4 and monitored by time-resolved UV-visible spectra. We have tested this reaction with the Fe3O4dpa@Pdx NPs catalysts, and the results are listed in Table 8. The measured TOFs are also very high, including that obtained with a sample with Pd deposited on its magnetite surface without the assistance of dpa (entry 1). In all cases, the reaction takes place with rapid release of H2 bubbles accompanied by a decrease of the absorption at 400 nm and the appearance of another at 317 nm, confirming the transformation of 4-nitrophenol to 4-aminophenol (Fig. 6a). The UV-vis spectra showed an isosbestic point (313 nm), indicating that no by-product is formed during the reduction of 4-NP. Since the concentration of NaBH4 exceeds that of 4-nitrophenol (c(NaBH4)/c(nitrophenol) = 100:1), the reduction can be considered as a pseudo-first-order reaction with regard to 4-nitrophenol only. The plot matches first-order reaction kinetics, and the rate constant is calculated from the equation ln(ct/c0) = kt. Fig. 6b shows the linear relationship between ln(ct/c0) and the reaction time for Fe3O4dpa@Pd0.3. For a quantitative comparison, we have introduced the activity parameter k′ = k(s−1)/mPd, where mPd is the total mass of the palladium added as catalyst. From the catalytic results (Table 8), it can be seen that the best sample is Fe3O4dpa@Pd0.1, which contains only 0.10% palladium. As commented above, this catalyst consists of Pd SACs, whose enormous catalytic activity has been recently recognized in different reactions.
 |
| | Scheme 3 Reduction of 4-NP. | |
Table 8 Activity of catalysts in 4-NP reduction
| Catalyst |
k′ (s−1 g−1) |
| Fe3O4@Pd |
24500 |
| Fe3O4dpa@Pd0.1 |
90000 |
| Fe3O4dpa@Pd0.3 |
58000 |
| Fe3O4dpa@Pd0.5 |
50600 |
| Fe3O4dpa@Pd1.0 |
16300 |
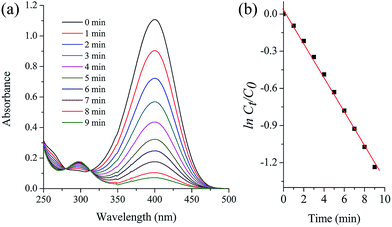 |
| | Fig. 6 Catalytic reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) with Fe3O4dpa@Pd0.3. (a) UV-vis spectra of the time dependent reaction. (b) lnCt/C0vs. time of reaction. | |
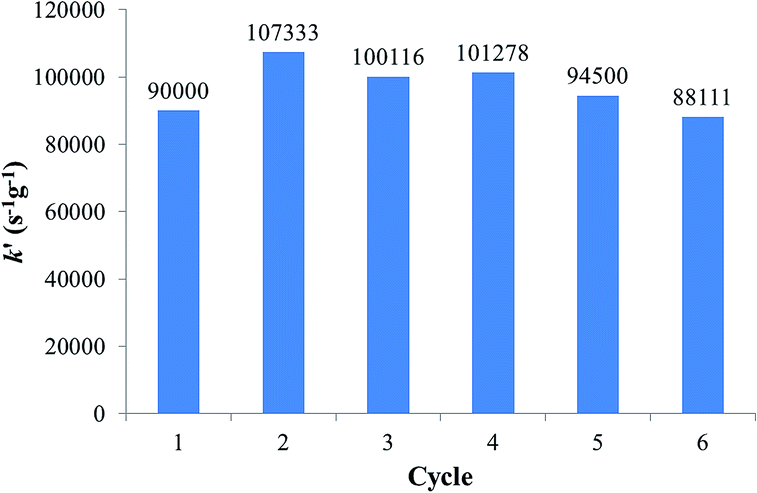
Table 9 permits us to compare the efficiency of numerous catalytic species, and it can be seen that the efficiency of the Fe3O4dpa@Pd0.1 NPs surpasses most of them. The reusability of the catalysts has been examined. The catalytic efficiency was maintained during the first six cycles for Fe3O4dpa@Pd0.1 (Fig. 7). Then, it began to decrease. ICPoes analysis indicated that the Pd content of the resulting NPs after six cycles decreased by only 6%. This fact, along with the constant activity shown for the catalyst through the cycles, seems to indicate that the reduction of nitrophenol occurs heterogeneously.
Table 9 Comparison of reduction of 4-NP catalyzed by Pd NPs from the literature
| Catalyst |
k (s−1) |
k′ (s−1 g−1) |
Ref. |
| Fe3O4dpa@Pd0.1 |
3.6 × 10−3 |
107333 |
This work |
| rGO/Pd–Fe3O4/PPy |
3.2 × 10−3 |
152153 |
37
|
| MOF |
1.2 × 10−2 |
7827 |
38
|
| [Fe3O4Sdp@Pd]ox |
4.4 × 10−2 |
6100 |
12
|
| PdNPs/Fe3O4/dopamine–phosphine |
2.0 × 10−2 |
2349 |
11a
|
| PdNPs/Fe3O4–Ag core–shell |
3.3 × 10−2 |
1736 |
39
|
| PdNPs/polypyrrole capsule |
8.9 × 10−3 |
1415 |
40
|
| PdNPs/fibrous nano-silica |
8.0 × 10−3 |
1026 |
41
|
| PdNPs/mesoporous silica |
1.2 × 10−2 |
750 |
42
|
| Pd-AuNPs/grapheme |
6.5 × 10−3 |
130 |
43
|
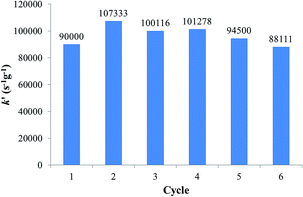 |
| | Fig. 7 Activity of Fe3O4dpa@Pd0.1 per cycle in 4-NP reduction. | |
Hydrogenation of styrene.
Very recently, we have described the catalytic activity of Fe3O4dopPPh2@Pdx NPs supported on magnetite nanoparticles using styrene hydrogenation (Scheme 4) as a model catalytic reaction in isopropanol as solvent at room temperature in a hydrogen atmosphere.17 Surprisingly, we observed that a sample containing a very low Pd weight content (0.18 wt%) and constituted uniquely of Pd SACs was absolutely inactive for the hydrogenation of styrene and other alkenes. This is the first time that Pd single atom catalysts (SACs) were shown to be catalytically inactive. In contrast, samples with higher Pd content, exhibiting small clusters and nanoparticles, gave exceptional catalytic results. These findings prompted us to analyse the catalytic activity of Fe3O4dpa@Pdx NPs for the hydrogenation of styrene in order to check whether our catalysts are able to mimic the catalytic behaviour shown by the Fe3O4dopPPh2@Pdx NPs. The results are compiled in Table 10. The most remarkable result was the null activity shown by the Fe3O4dpa@Pd0.1 NPs (entry 2). This result confirms the inactivity of Pd SACs for the hydrogenation of styrene, and it is in contrast to the excellent catalytic efficiency reported in a number of processes.44 Significantly, the remaining samples followed the expected catalytic trend, that is, excellent performances that deteriorated with increasing Pd content and Pd nanoparticle size.
 |
| | Scheme 4 Styrene hydrogenation. | |
Table 10 Activity of the catalyst in styrene hydrogenationa
| Entry |
Catalyst |
TOF (h−1) |
|
3 bar H2, 5 mmol styrene, 25 °C, EtOH, 1 h.
|
| 1 |
Fe3O4@Pd |
404 |
| 2 |
Fe3O4dpa@Pd0.1 |
0 |
| 3 |
Fe3O4dba@Pd0.3 |
4690 |
| 4 |
Fe3O4dba@Pd0.5 |
4880 |
| 5 |
Fe3O4dba@Pd1.0 |
3630 |
Reusability studies have been carried out on the best catalytic sample, Fe3O4dpa@Pd0.5. The loss of activity is very small (Table 11); this is confirmed by the final Pd content after 3 rounds, which is less than 3% in relation to the starting sample. These results strongly support heterogeneous catalytic behaviour for this process. Table 12 lists selected catalysts for the hydrogenation of styrene, including reaction conditions and TOFs; the Pd catalysts reported here are among the best examples.
Table 11 Styrene hydrogenation per cycle with Fe3O4dpa@Pd0.5 as catalysta
| Cycle |
TOF (h−1) |
|
3 bar H2, 5 mmol styrene, 25 °C, EtOH.
|
| 1 |
4880 |
| 2 |
4793 |
| 3 |
4760 |
Table 12 Comparison of styrene hydrogenation catalyzed by Pd NPs from the literature
| Catalyst |
Solvent |
Pressure (bar) |
T (°C) |
TOF (h−1) |
Ref. |
| Fe3O4dpa@Pd0.5 |
EtOH |
3 |
25 |
4880 |
This work |
| [Fe3O4Sdp@Pd]ox |
EtOH |
3 |
25 |
3100 |
12
|
| Polivinilpiridina/PdCl2 |
EtOH |
1 |
25 |
6944 |
45
|
| PdNPs/polietilenglicol |
EtOH |
3 |
25 |
660 |
46
|
| Polyestyrene/Pd complex |
DMF |
1 |
25 |
766 |
47
|
| PdNPs/stearate/oleyl amine |
Cyclohexane |
21 |
50 |
7704 |
48
|
| MOF/Pd |
None |
1 |
35 |
703 |
49
|
| Pd/C |
Ethyl acetate |
3 |
25 |
163 |
50
|
Conclusions
In conclusion, in this work we demonstrate that the use of the dpa molecule as a linker for the deposition of palladium species on the surface of magnetite permitted partial solubilization of the resulting hybrid in water. This fact, along with the partial removal of dpa during the reduction process, facilitates the approach of the substrates to the catalyst. In addition to this, the small size of the Pd NPs supported on magnetite in Fe3O4dba@Pd0.3 and Fe3O4dba@Pd0.5, along with the presence of Pd SACs, explains the exceptional results for the Suzuki C–C coupling reaction and hydrogenation of 4-nitrophenol both in neat water and in a mixture of ethanol/water.
Experimental
All manipulations were performed under purified nitrogen using standard Schlenk techniques. Organic Elemental Analysis (OEA) was performed in an elemental analyzer, Thermo Scientific Flash 2000 A7 model. The infrared (IR) spectra were plotted with a NICOLET Impact 400 FT-IR. The abbreviations used for signals were w (weak), m (medium), and s (strong); only significant signals are mentioned. Thermogravimetric Analysis (TGA) was performed on an IGA 851 Mettler-Toledo instrument, nitrogen flow (50 mL min−1), from 30 °C to 1000 °C at a gradient of 10 °C min−1. ICPoes (Inductively Coupled Plasma optical emission spectrometry) analysis was achieved in a Perkin Elmer 3200RL model. Samples were dissolved in an HCl/HNO3 3:1 (v/v) mixture. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and energy-dispersive X-ray spectroscopy (EDS) were carried out using a double spherical aberration-corrected JEOL JEM-ARM200F microscope operated at 200 kV and equipped with a JEOL Dry SD100GV silicon drift detector with a 100 mm2 detection area for EDS analysis. The microscope was set up in STEM mode with a probe semiconvergence angle set to 25.3 mrad, which yields a calculated probe size of about 80 pm. The annular semidetection range of the annular dark-field detector was set to collect electrons scattered between 90 and 370 mrad. Transmission electron microscopy (TEM) images were registered at 200 kV using a JEOL 2010F instrument having a point resolution of 0.21 nm. Samples were prepared by placing a drop of solution on a holey-carbon-coated Cu TEM grid and allowing the solvent to evaporate in air. In addition, for the HAADF-STEM experiments, the samples were gently plasma cleaned with Ar (75%)/O2 (25%) plasma for a few seconds to remove hydrocarbon contamination from the surfaces of the nanoparticles. XPS (X-ray Photoelectron Spectroscopy) analysis was carried out in a SPECS instrument with an Al X-ray source and a Phoibos 150 analyzer. The spectrum was recorded at under 10−7 Pa. Binding energies were referred to the C 1s signal at 284.8 eV. All reactants were purchased from suppliers at synthetic quality or higher and were used without further purification. The solvents were HPLC quality and/or were dried using a pure solve instrument from Innovate Technology USA Inc. Deionized water was obtained from a Millipore Helix 3 water purification system.
Synthesis of ferrite nanoparticles (Fe3O4)
6.95 g (25.3 mmol) of iron(II) sulphate heptahydrate was mixed with 10.0 g of iron(III) sulphate hydrate and dissolved in water (250 mL). Then, 25% ammonia solution was slowly added to adjust the pH to 10. The reaction mixture was stirred for 1 h at 50 °C. The nanoparticles obtained were separated magnetically and washed with water until pH 7 was attained. The resulting nanoparticles were dried under reduced pressure at 60 °C for 2 h.
Synthesis of Fe3O4@Pd
100 mg of magnetite nanoparticles were sonicated in methanol (10 mL) for 30 min. Then, 4.1 mg (12.5 μmol) of K2[PdCl4] dissolved in water (2 mL) were added and stirred (1000 rpm) for 2 h. After that, the nanoparticles were washed with water (3 × 10 mL), acetone (10 mL), and water (10 mL). Next, the NPs were dispersed in water (8 mL) and an aqueous solution (2.5 mL) of sodium borohydride (0.05 M) was added. Then, the nanoparticles were washed with water (3 × 30 mL) and acetone, removed by an external magnet and dried under reduced pressure. ICPoes: 0.68% Pd.
Synthesis of Fe3O4dpa
400 mg of ferrite nanoparticles were sonicated in ethanol (15 mL) for 30 min. Then, a solution of 4-(diphenylphosphino)benzoic acid (dpa, 306 mg, 1 mmol) in methanol (15 mL) was added to the suspended Fe3O4. This mixture was sonicated for 2 h, and the resulting nanoparticles were washed with ethanol (3 × 30 mL) and acetone (30 mL). Finally, the Fe3O4dpa nanoparticles were collected by magnetic separation and dried under reduced pressure. OEA: 2.4% C, 3.2% dpa.
Synthesis of Fe3O4dpa@Pdx (x = 1, 0.5, 0.3, 0.1)
400 mg of Fe3O4dpa were dispersed in water (40 mL) for 10 min by sonication. Next, a quantity (depending on x; 50 μmol, 25 μmol, 12 μmol, or 4 μmol, respectively) of K2[PdCl4] dissolved in water (8 mL) was added and stirred (1000 rpm) for 2 h. After that, the nanoparticles were washed with water (1 × 50 mL), ethanol (2 × 50 mL) and acetone (50 mL), and dried under reduced pressure. Next, 400 mg of these nanoparticles were dispersed in water (40 mL) by sonication, and 10 mL of an aqueous solution of sodium borohydride (0.05 M) was added. The mixture was stirred (1200 rpm) for 2 h. Then, the nanoparticles were washed with water (1 × 50 mL), ethanol (2 × 50 mL) and acetone (50 mL), removed by an external magnet and dried under reduced pressure. ICPoes: 1.1%, 0.50%, 0.32%, and 0.097% of palladium for Fe3O4dpa@Pdx, where x = 1, 0.5, 0.3, and 0.1, respectively.
Suzuki–Miyaura coupling
In a typical Suzuki–Miyaura reaction, 3.6 mmol of phenylboronic acid, 9 mmol of base, and a quantity of Pd catalyst (e.g. 2.0 mg of Fe3O4Sdp@Pd0.5, 9.4 × 10−5 mmol) were weighed into a Schlenk tube. The mixture was purged with nitrogen and 60 mL of solvent was added. After that, the tube was brought to a preheated plate at 65 °C, and 3 mmol (1 eq.) of substrate was added. When the desired reaction time was reached, the mixture was allowed to cool. Extraction with ethyl acetate was performed. The organic phase was removed, dried over sodium sulphate and analyzed by GC. The identity of the products was confirmed by GC-MS analysis.
Catalytic reduction of p-nitrophenol
30 μL of p-nitrophenol (7.4 mM) and 30 μL of NaBH4 (0.40 M) were added to a quartz cuvette containing 2 mL of water. Then, 30 μL of an aqueous suspension containing the catalyst nanocomposite (approximately 3 to 5 mg in 5 mL of water) was injected into the cuvette to start the reaction. The intensity of the absorption peak at 400 nm in the UV-vis spectra was used to monitor the process of the conversion of p-nitrophenol to p-aminophenol. After each cycle of reaction, another 30 μL of p-nitrophenol and 30 μL of NaBH4 were added to the reaction to study the reuse of the catalyst. The catalytic reduction reactions were conducted at room temperature.
Hydrogenation of styrene
Hydrogenation reactions were carried out in a hydrogen atmosphere at room temperature. Typically, under nitrogen, ferrite catalyst (5.0 mg approximately) was dispersed in freshly distilled ethanol (20 mL), and then styrene (2.5 mmol) was added by pouring. After that, the mixture was transferred into a Fisher–Porter reactor, filled with hydrogen (3 bar), and stirred at constant speed. The conversion was determined by GC analysis.
Acknowledgements
This work was financially supported by the MICINN (projects CTQ2012-31335 and CTQ2015-65040-P). J. L. is a Serra Húnter Fellow and is grateful to the ICREA Academia program. Access to the TEM facilities at IBM Research-Zurich, Switzerland, under the IBM/Empa Master Joint Development Agreement is gratefully acknowledged. A. G. thanks the University of Tolima (Colombia) for financial support.
References
-
(a) M. Lamblin, L. Nassar-Hardy, J. C. Hierso, E. Fouquet and F. X. Felpin, Adv. Synth. Catal., 2010, 352, 33–79 CrossRef CAS;
(b) S. Paul, M. M. Islam and S. M. Islam, RSC Adv., 2015, 5, 42193–42221 RSC.
- F. Lu, J. Ruiz and D. Astruc, Tetrahedron Lett., 2004, 45, 9443–9445 CrossRef CAS.
- A. Balanta, C. Godard and C. Claver, Chem. Soc. Rev., 2011, 40, 4973–4985 RSC.
- R. M. Crooks, M. Zhao, L. Sun, V. Chechik and L. K. Yeung, Acc. Chem. Res., 2000, 34, 181–190 CrossRef.
- V. K. R. Kumar and K. R. Gopidas, Tetrahedron Lett., 2011, 52, 3102–3105 CrossRef.
- E. Raluy, I. Favier, A. M. López-Vinasco, C. Pradel, E. Martin, D. Madec, E. Teuma and M. Gómez, Phys. Chem. Chem. Phys., 2011, 13, 13579–13584 RSC.
- P. Sonström and M. Bäumer, Phys. Chem. Chem. Phys., 2011, 13, 19270–19284 RSC.
-
(a) D. Wang and D. Astruc, Chem. Rev., 2014, 114, 6949–6985 CrossRef CAS PubMed;
(b) R. B. N. Baig and R. S. Varma, Chem. Commun., 2013, 49, 752–770 RSC;
(c) W. Dong and D. Astruc, Molecules, 2014, 19, 4635–4653 CrossRef PubMed;
(d) W. Dong, C. Deraedt, L. Salmo, C. Labrugere, L. Etienne, J. Ruiz and D. Astruc, Chem.–Eur. J., 2014, 21, 6501–6512 Search PubMed;
(e) L. M. Rossi, N. J. S. Costa, F. P. Silva and R. Wojcieszak, Green Chem., 2014, 16, 2906–2933 RSC;
(f) M. B. Gawande, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 3371–3393 RSC;
(g) S. Roy and M. A. Pericàs, Org. Biomol. Chem., 2009, 7, 2669–2677 RSC.
-
(a) R. Polshettivar, R. Luque, A. Fihri, H. Zhu, M. Bouhra and J. M. Basset, Chem. Rev., 2011, 111, 3036–3075 CrossRef PubMed;
(b) V. Polshettiwar and R. S. Varma, Green Chem., 2010, 12, 743–754 RSC;
(c) R. B. N. Baig and R. S. Varma, Chem. Commun., 2012, 48, 2582–2584 RSC;
(d) R. B. N. Baig and R. S. Varma, Green Chem., 2013, 15, 398–417 RSC.
-
(a) K. Jiang, H.-X. Zhang, Y.-Y. Yang, R. Mothes, H. Lang and W.-B. Cai, Chem. Commun., 2011, 43, 11924–11926 RSC;
(b) P. Li, L. Wang, L. Zhang and G.-W. Wang, Adv. Synth. Catal., 2012, 354, 1307–1318 CrossRef CAS.
-
(a) F. Gonzàlez de Rivera, I. Angurell, M. D. Rossell, R. Erni, J. Llorca, N. J. Divins, G. Muller, M. Seco and O. Rossell, Chem.–Eur. J., 2013, 19, 11963–11974 CrossRef PubMed;
(b) N. J. S. Costa, P. K. Kiyohara, A. L. Monteiro, Y. Coppel, K. Philippot and L. M. Rossi, J. Catal., 2010, 276, 382–389 CrossRef CAS.
- A. Guarnizo, I. Angurell, M. D. Rossell, J. Llorca, G. Muller, M. Seco and O. Rossell, RSC Adv., 2015, 5, 91340–91348 RSC.
- J. M. Thomas, Nature, 2015, 525, 325–326 CrossRef CAS PubMed.
- V. Polshettiwar and R. S. Varma, Chem.–Eur. J., 2009, 15, 1582–1586 CrossRef CAS PubMed.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- G. S. Parkinson, Z. Novotny, G. Argentero, M. Schmid, J. Pavelec, R. Kosak, P. Blaha and U. Diebold, Nat. Mater., 2013, 12, 724–728 CrossRef CAS PubMed.
- M. D. Rossell, F. J. Caparrós, I. Angurell, G. Muller, J. Llorca, M. Seco and O. Rossell, Catal. Sci. Technol., 2016, 6, 4081–4086 CAS.
-
(a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS;
(b) M. Moreno-Mañas and R. Pleixats, Acc. Chem. Res., 2003, 36, 638–643 CrossRef PubMed;
(c) A. Suzuki and Y. Yamamoto, Chem. Lett., 2011, 40, 894–901 CrossRef CAS;
(d) A. Balanta, C. Godard and C. Claver, Chem. Soc. Rev., 2011, 40, 4973–4985 RSC.
- A. Bej, K. Ghosh, A. Sarkar and D. W. Knight, RSC Adv., 2016, 6, 11446–11453 RSC.
-
(a) M. J. Jin and D. H. Lee, Angew. Chem., Int. Ed., 2010, 49, 1119–1122 CrossRef CAS PubMed;
(b) A. Taher, J. B. Kim, J. Y. Jun, W. S. Ahn and M. J. Jin, Synlett, 2009, 2477–2482 CAS;
(c) Z. Yinghuai, S. C. Peng, A. Emi, S. Zhenshun, M. Lisa and R. A. Kemp, Adv. Synth. Catal., 2007, 349, 1917–1922 CrossRef;
(d) B. Karimi, F. Mansouri and H. Vali, Green Chem., 2014, 16, 2587–2596 RSC.
- D. Rosario-Amorin, X. Wang, M. Gaboyard, R. Clérac, S. Nlate and K. Heuzé, Chemistry, 2009, 15, 12636–12643 CrossRef CAS PubMed.
- Q. Du, W. Zhang, H. Ma, J. Zheng, B. Zhou and Y. Li, Tetrahedron, 2012, 68, 3577–3584 CrossRef CAS.
- P. Li, L. Wang, L. Zhang and G.-W. Wang, Adv. Synth. Catal., 2012, 354, 1307–1318 CrossRef CAS.
- J. Sun, Y. Fu, G. He, X. Sun and X. Wang, Appl. Catal., B, 2015, 165, 661–667 CrossRef CAS.
- F. Chahdoura, I. Favier, C. Pradel, S. Mallet-Ladeira and M. Gómez, Catal. Commun., 2015, 63, 47–51 CrossRef CAS.
- D. Guin, B. Baruwati and S. V. Manorama, Org. Lett., 2007, 9, 1419–1421 CrossRef CAS PubMed.
- L. Wu, B.-L. Li, Y.-Y. Huang, H.-F. Zhou, Y.-M. He and Q.-H. Fan, Org. Lett., 2006, 8, 3605–3608 CrossRef CAS PubMed.
- R. Cano, D. J. Ramón and M. Yus, Tetrahedron, 2011, 67, 5432–5436 CrossRef CAS.
- C. Deraedt, L. Salmon, L. Etienne, J. Ruiz and D. Astruc, Chem. Commun., 2013, 49, 8169–8171 RSC.
- B. Karimi, F. Mansouri and H. Vali, Green Chem., 2014, 16, 2587–2596 RSC.
- F. Yang, C. Chi, S. Dong, C. Wang, X. Jia, L. Ren, Y. Zhang, L. Zhang and Y. Li, Catal. Today, 2015, 256, 186–192 CrossRef CAS.
- S. Su, G. Yue, D. Huang, G. Yang, X. Lai and P. Zhao, RSC Adv., 2015, 5, 44018–44021 RSC.
- T. Aditya, A. Pal and T. Pal, Chem. Commun., 2015, 51, 9410–9431 RSC.
- H. Wei, X. Liu, A. Wang, L. Zhang, B. Qiao, X. Yang, Y. Huang, S. Miao, J. Liu and T. Zhang, Nat. Commun., 2014, 5, 5634 CrossRef CAS PubMed.
- G. Vilé, D. Albani, M. Nachtegaal, Z. Chen, D. Dontsova, M. Antonietti, N. Lopez and J. Perez-Ramirez, Angew. Chem., Int. Ed., 2015, 54, 11265–11269 CrossRef PubMed.
- K. Hayakawa, T. Yoshimura and K. Esumi, Langmuir, 2003, 19, 5517–5521 CrossRef CAS.
- T. Yao, H. Wang, Q. Zuo, J. Wu, X. Zhang, F. Cui and T. Cui, Chem.–Asian J., 2015, 10, 1940–1947 CrossRef CAS PubMed.
- Z. Dong, X. Le, Y. Liu, C. Dong and J. Ma, J. Mater. Chem. A, 2014, 2, 18775–18785 CAS.
- K. Jiang, H.-X. Zhang, Y.-Y. Yang, R. Mothes, H. Lang and W.-B. Cai, Chem. Commun., 2011, 47, 11924–11926 RSC.
- Y. Xue, X. Lu, X. Bian, J. Lei and C. Wang, J. Colloid Interface Sci., 2012, 379, 89–93 CrossRef CAS PubMed.
- X. Le, Z. Dong, X. Li, W. Zhang, M. Le and J. Ma, Catal. Commun., 2015, 59, 21–25 CrossRef CAS.
- J. Morère, M. J. Tenorio, M. J. Torralvo, C. Pando, J. A. R. Renuncio and A. Cabañas, J. Supercrit. Fluids, 2011, 56, 213–222 CrossRef.
- C.-H. Liu, R.-H. Liu, Q.-J. Sun, J.-B. Chang, X. Gao, Y. Liu, S.-T. Lee, Z.-H. Kang and S.-D. Wang, Nanoscale, 2015, 7, 6356–6362 RSC.
-
(a) E. J. Peterson, A. T. De La Riva, S. Lin, R. S. Johnson, H. Guo, J. T. Miller, J. H. Kwak, C. H. F. Peden, B. Kiefer, L. F. Allard, F. H. Ribeiro and A. K. Datye, Nat. Commun., 2014, 5, 4885 CrossRef CAS PubMed;
(b) S. F. J. Hackett, R. M. Brydson, M. H. Gass, I. Harvey, A. D. Newman, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2007, 46, 8593–8596 CrossRef CAS PubMed;
(c) H. Yan, H. Cheng, H. Yi, Y. Lin, T. Yao, C. Wang, J. Li, S. Wei and J. Lu, J. Am. Chem. Soc., 2015, 137, 10484–10487 CrossRef CAS PubMed;
(d) G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Science, 2012, 335, 1209–1212 CrossRef CAS PubMed.
- S. Gao, W. Li and R. Cao, J. Colloid Interface Sci., 2015, 441, 85–89 CrossRef CAS PubMed.
- F. A. Harraz, S. E. El-Hout, H. M. Killa and I. A. Ibrahim, J. Catal., 2012, 286, 184–192 CrossRef CAS.
- S. M. Islam, A. S. Roy, P. Mondal and N. Salam, Appl. Organomet. Chem., 2012, 26, 625–634 CrossRef CAS.
- C. H. Yen, H.-H. Wei, H.-W. Lin and C.-S. Tan, Appl. Organomet. Chem., 2012, 26, 736–742 CrossRef CAS.
- Y. Pan, D. Ma, H. Liu, H. Wu, D. He and Y. Li, J. Mater. Chem., 2012, 22, 10834–10839 RSC.
- C.-B. Hwang, Y.-S. Fu, Y.-L. Lu, S.-W. Jang, P.-T. Chou, C. R. C. Wang and S. J. Yu, J. Catal., 2000, 195, 336–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XPS of catalysts (Fig. S1), tables of conversion in Suzuki–Miyaura based on temperature (in ethanol/water) (Table S1), base (Table S2), solvent (Table S3) and temperature in water solution (Table S4). See DOI: 10.1039/c6ra14257e |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a