
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the structural relationship between encapsulated antimicrobial peptides and the bilayer membrane mimetic lipidic cubic phase: studies with gramicidin A′†
Thomas G.
Meikle
ab,
Charlotte E.
Conn
c,
Frances
Separovic
a and
Calum J.
Drummond
*bc
aSchool of Chemistry, Bio21 Institute, University of Melbourne, VIC 3010, Australia
bCSIRO Manufacturing Flagship, Bag 10, Clayton, VIC 3169, Australia. E-mail: calum.drummond@rmit.edu.au
cSchool of Science, College of Science, Engineering and Health, RMIT University, Melbourne, VIC 3000, Australia
First published on 19th July 2016
Abstract
Lipid based bicontinuous cubic mesophases provide a low-cost, robust membrane mimetic nanomaterial which allows for the incorporation of membrane peptides and proteins. However, the relationship between the mesostructure of the host lipidic bicontinuous mesophase, the chemical structure of its constituents, and the secondary structure of encapsulated biomolecules is complex, and at present largely unclear. Here we have examined the effects of adding gramicidin A′, an anti-microbial peptide, to lipidic bicontinuous cubic phases composed of a number of different lipids. We demonstrate, using a combination of synchrotron small angle X-ray scattering and circular dichroism, that fundamental physicochemical parameters of the lipid mesophase impact both the structural response to peptide addition, and the conformation of the encapsulated peptide. We have rationalised the results with reference to hydrophobic mismatch, the putative lateral pressure profile and the intrinsic surface curvature of each lipid system. Results should be of use for several applications of hybrid peptide-lipid materials including peptide based drug delivery and the design of in meso crystallization trials.
Introduction
Self-assembled lipidic bicontinuous cubic mesophases provide an ideal medium for the incorporation of hydrophilic, hydrophobic and amphiphilic peptides and proteins.1 They are additionally recognized as providing an inexpensive and robust model bilayer and cell membrane mimetic with a number of properties that provide an advantage over other bilayer structures. Here we used the inverse bicontinuous cubic mesophase to examine the peptide–bilayer interactions resulting from incorporation of the peptide gramicidin A′ (gA′). Generalized observations are of interest for practical applications, such as in meso crystallization,2 the design of drug delivery materials and biosensors,1,3–5 as well as providing fundamental knowledge on the interactions of peptides with membranes.Bicontinuous cubic phases consist of a lipid bilayer, curved in three-dimensional space into the shape of a triply periodic minimal surface. Three bicontinuous cubic structures are experimentally observed in pure lipid systems, including the Schwarz diamond (QDII with crystallographic space group Pn3m), the primitive (QPII with space group Im3m) and the Schoen gyroid (QGII with space group Ia3d).6 The bilayer in each of these cubic mesophases features zero mean curvature and a high surface area to volume ratio, and results in a network of two interpenetrating but unconnected water channels (Fig. 1).
 | ||
| Fig. 1 3D representation of the cubic Im3m mesophase, with the lipid bilayer shown in red, and the two interpenetrating water channels in blue and green. The bilayer is removed on the right side to clearly show the water channel structure. Modelled with K3Dsurf and Blender.7,8 | ||
Lipidic bicontinuous cubic mesophases offer a range of advantages for the incorporation of proteins and peptides. The properties of lipidic cubic mesophases are comparable to those existing in biological membranes, and thus encapsulated proteins and peptides can potentially retain their activity. These properties make the lipidic cubic phase a highly prospective material for drug delivery applications,5,9 including the encapsulation and controlled release of therapeutic proteins and peptides within the body. They are particularly useful for hydrophobic or amphiphilic molecules not suited to traditional administration routes. A number of proteins and peptides have demonstrated controlled release from lipidic cubic mesophases.10,11 Complex, triggered release materials are possible,12 whereby proteins encapsulated in the bilayer function to increase and decrease the rate of drug release via an opening and closing of pores or channels. The lipidic cubic phase can also be used to probe fundamental peptide–lipid interactions.
Guest biomolecules, when inserted into the bilayer, can influence the symmetry and structural unit dimensions of lipidic cubic mesophases.13 If we are to take full advantage of these materials we must understand the complex relationship between the chemical composition of the bilayer, the lipidic mesostructure, and the guest biomolecule. We have studied the effects of the antimicrobial peptide gA′ on cubic phases comprised of a range of commercially available lipids, tracking the changes over a period of two days. The evolution of the cubic structure as the concentration of peptide was increased was correlated with both the structure of the host lipid and the structure of gA′ within the bilayer.
Gramicidin A′ is a mixture of the linear pentadecapeptides gramicidin A, B and C; comprising ∼80%, ∼6% and ∼14%, respectively (Fig. 2). This antimicrobial peptide is produced by Bacillus brevis and exerts its effect by forming a dimeric channel spanning the bilayer and facilitating the uncontrolled movement of monovalent cations (Na+) between the intra- and extra-cellular environment, leading to cell death. Gramicidin A′ is well studied,14–16 cheap, stable, and structure grade crystals have been grown in meso,17 indicating that the peptide is readily reconstituted in the lipidic cubic phase. In addition, pore forming peptides have applications as drugs18 and can be used to further our understanding of a range of diseases where the formation of pores in the cell membrane are believed to play a role, including Alzheimer's disease19 and bacterial infections.20
 | ||
| Fig. 2 Structural diagram showing gramicidin A, B and C, both valine and isoleucine variants. | ||
Since its sequence elucidation in 1964,21 there has been extensive work to understand the function and structure of gA′. The peptide can exist as a variety of different conformations depending on its local environment22 and forms two major folding types: the double helix and the helical dimer (Fig. 3). The double helix, also termed the “pore” conformation is the result of two monomers forming a series of intermolecular hydrogen bonds along their length in either a parallel or anti-parallel β-sheet like motif, forming a double helix. This is the dominant motif present in solution in most organic solvents,23 and usually consists of four interconverting conformational variant species at equilibrium.
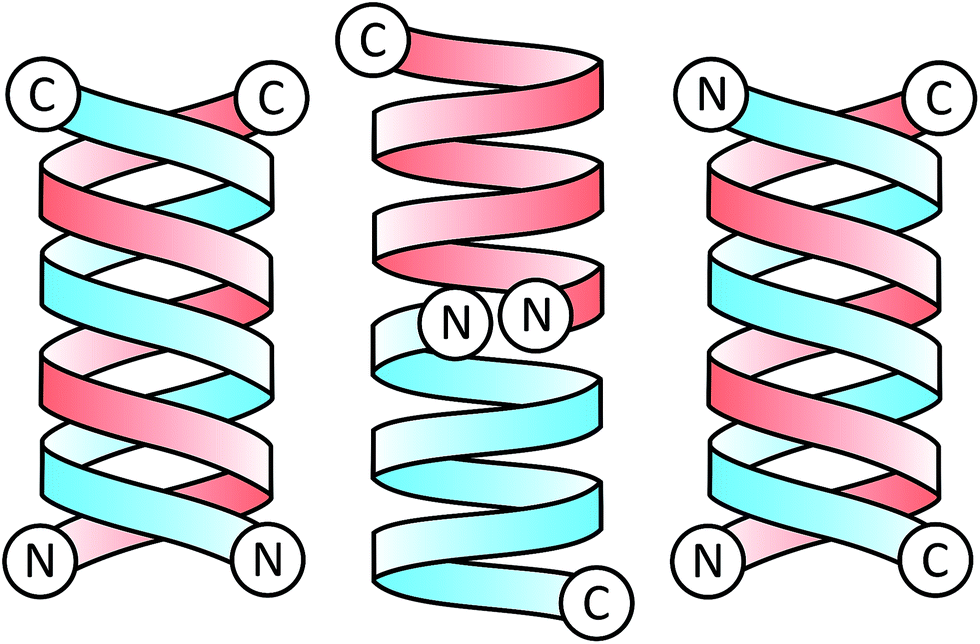 | ||
| Fig. 3 Schematic illustration of gramicidin A dimers in the parallel double helix form (left), helical dimer form (centre), and anti-parallel double helix form (right). Each of the identical gramicidin monomers comprising the dimer is highlighted in either red or blue for clarity, with C and N representing the C and N termini. | ||
The helical dimer or “channel” conformation results from a series of intramolecular hydrogen bonds occurring along the length of the molecule which warps to form a single helical monomer. The dimer is formed when two of these monomers are joined anti-parallel, N-terminal to N-terminal through 6 intermolecular hydrogen bonds linking the formyl groups and the N-terminal amino acids with one another. This conformation is also referred to as a β6.3 structure,24 due to the 6.3 residues per turn in the helix.
Caffrey et al. has demonstrated that addition of gA′ to a series of cubic phase monoacylglycerol (MAG) lipids results in a change in symmetry, an effect closely correlated with hydrophobic mismatch.25 Herein we report the effect of peptide addition to a less studied class of cubic-phase forming lipids, the branched chain or isoprenoid-type lipids. These include phytantriol (PT), a saturated branched chain lipid frequently used as a cosmetic ingredient, and phytanoyl monoethanolamide (PE), a custom lipid synthesized in-house and designed to maintain cubic symmetry at low temperatures suitable for in meso crystallization.26 Similar lipids are found in archaebacteria and show enhanced stability compared to MAGs. The branched hydrocarbon chain architecture has been shown to result in increased local lateral pressure when compared to equivalent straight chain lipids.27 In addition, we expand upon previous studies on the effect of hydrophobic mismatch using MAG lipids by including another MAG, monovaccenin (MV), in addition to monoolein (MO) and monopalmitolein (MP). We have investigated a mixture of monoolein + 8 wt% cholesterol, an important biomolecule which, at this concentration, has been found to facilitate crystal growth in meso. These experiments were designed to explore the mesophase behaviour of a variety of commonly used cubic phase lipids subsequent to peptide addition, and to compare between two distinct classes of lipid: the monoacylglycerols and the branched chain isoprenoids. For direct relevance to applications such as in meso crystallization and drug delivery, we have varied the lipid between predominately commercially available lipids (MO, MV, MP, PT), and one custom synthesized lipid (PE).
Experimental section
Materials
Monoolein, cholesterol and gramicidin A′ (sold as gramicidin D for Dubos) were purchased from Sigma-Aldrich (St Louis, MO). Monovaccenin and monopalmitolein were purchased from Nu Chek Prep (Elysian, MN). 3,7,11,15-Tetramethyl-1,2,3-hexadecanetriol (phytantriol) (96%) was provided by DSM Nutritional Products (Herleen, Netherlands). Phytanoyl monoethanolamide (PE) was synthesized in house, following the synthesis outlined in a previous publication.26 SD-2 96-well crystallisation plates were purchased from IDEX Corp. (Rohnert Park, CA).Plate set-up
Synchrotron SAXS experiments were conducted in SD-2 96-well crystallization plates, which feature two subwells and a single reservoir for each plate coordinate. Each lipid was hydrated to 80% w/w water; all lipids formed the QDII cubic phase under excess water conditions. The high water content prevents dehydration experienced by sealed plates during equilibration and transport. Gramicidin A′ was added to each of the six host lipids at eight concentrations ranging 0.1 to 15 mol%, with mol% defined as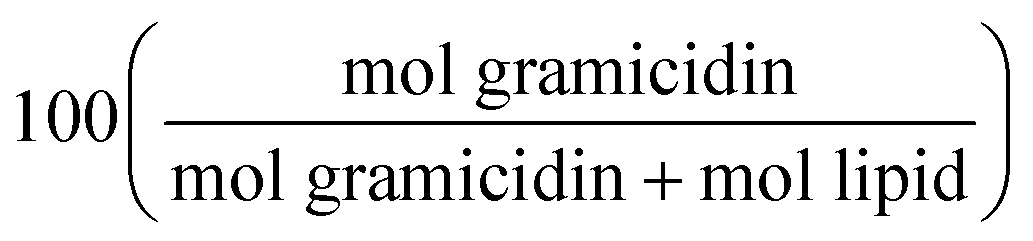 . The mixture of MO + 8 wt% cholesterol was assumed to possess the same molecular weight as pure MO in all calculations. Plates were measured on the SAXS beamline at the Australian Synchrotron at four different time points: 2, 12, 24 and 48 hours after initial preparation.
. The mixture of MO + 8 wt% cholesterol was assumed to possess the same molecular weight as pure MO in all calculations. Plates were measured on the SAXS beamline at the Australian Synchrotron at four different time points: 2, 12, 24 and 48 hours after initial preparation.
Gramicidin A′, dissolved in either methanol or ethanol at 8 different concentrations (one for each final gramicidin A′ concentration), was dispensed into each well of a 96-well plate using a Mosquito robot (TTP Labtech Ltd, Melbourn, UK). Note that due to robotic protocols a set volume of each peptide stock was used for all lipids, such that the final mol% value differs slightly for each lipid. Lipids were dissolved in ethanol to a concentration of 200 mg mL−1 and 1.05 μL was deposited in each well over the peptide giving a lipid mass of 0.21 mg. The plate was then dried for 5 days under vacuum to remove all solvent. The dry plates were hydrated at various times prior to SAXS screening by dispensing 0.84 μL of water into each well, resulting in a solvent to lipid ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20. Lastly, 20 μL of water was added to the reservoir of each well and the plate sealed.
:20. Lastly, 20 μL of water was added to the reservoir of each well and the plate sealed.
Synchrotron small-angle X-ray scattering measurements
SAXS data were obtained using the SAXS/WAXS beamline at the Australian Synchrotron. A typical SAXS experiment used a beam of wavelength λ = 1.23984 Å (10.000 ± 0.002 keV) with dimensions 250 μm × 120 μm with a typical flux of 5 × 1012 photons per s. The distance from sample to detector (Pilatus 1-M) was 900 mm. The custom-built plate holder accommodates 2 plates of standard SBS footprint (85 × 127 mm, 96 or 384 well) and is mounted directly in front of the beam. Temperature control is achieved with fan-forced air temperature control. The air is forced over a Peltier controlled heat exchanger, backed by a water cooled/heated plate running from a water bath, to maintain the samples at a constant temperature of 20 °C. The plates are loaded and mounted in front of the beam, coordinates of the samples mapped and an automated scan of each well is initiated. Using an exposure time of 1 s, all 192 samples in both 96 well plates can be screened in ∼13 min.Data analysis
Ordered lyotropic liquid crystalline mesophases produce distinct diffraction patterns when subjected to high intensity X-ray beams (Fig. 4), which allow determination of the lipid phase given adequate resolution. A new high-throughput analysis program, RapidPhaseIdent, developed by Dr Stephen Mudie (Australian Synchrotron) was used to analyse the large high-throughput data sets generated. 2D diffraction images are initially converted to 1D files of intensity vs. q using the Scatterbrain program. The RapidPhaseIdent program selects diffraction peaks within a pre-set q-range for each image. The program then selects the most likely mesophase from a pre-set list of common lipid mesophases including the cubic mesophases. RapidPhaseIdent has been used successfully to analyse large SAXS data sets that were generated in a similar fashion.28 The lattice parameter (and error) for each image can be exported as a .csv file. RapidPhaseIdent was used in conjunction with the IDL-based AXCESS software package, developed at Imperial College London,29 which allows for individual analysis of the X-ray images. Using AXCESS, 2D images of diffraction patterns are integrated and a 1D diffraction plot of intensity vs. pixel number is produced. After calibrating the program with a known standard (silver behenate, d = 58.38 Å), and assigning the sample peaks, the program calculates the d-spacing for a given phase. The program specifies a set of adjustable guidelines for each of the commonly observed phase symmetries, which assists with phase determination. Where two or more phases are found to coexist, the d-spacing of each is calculated separately.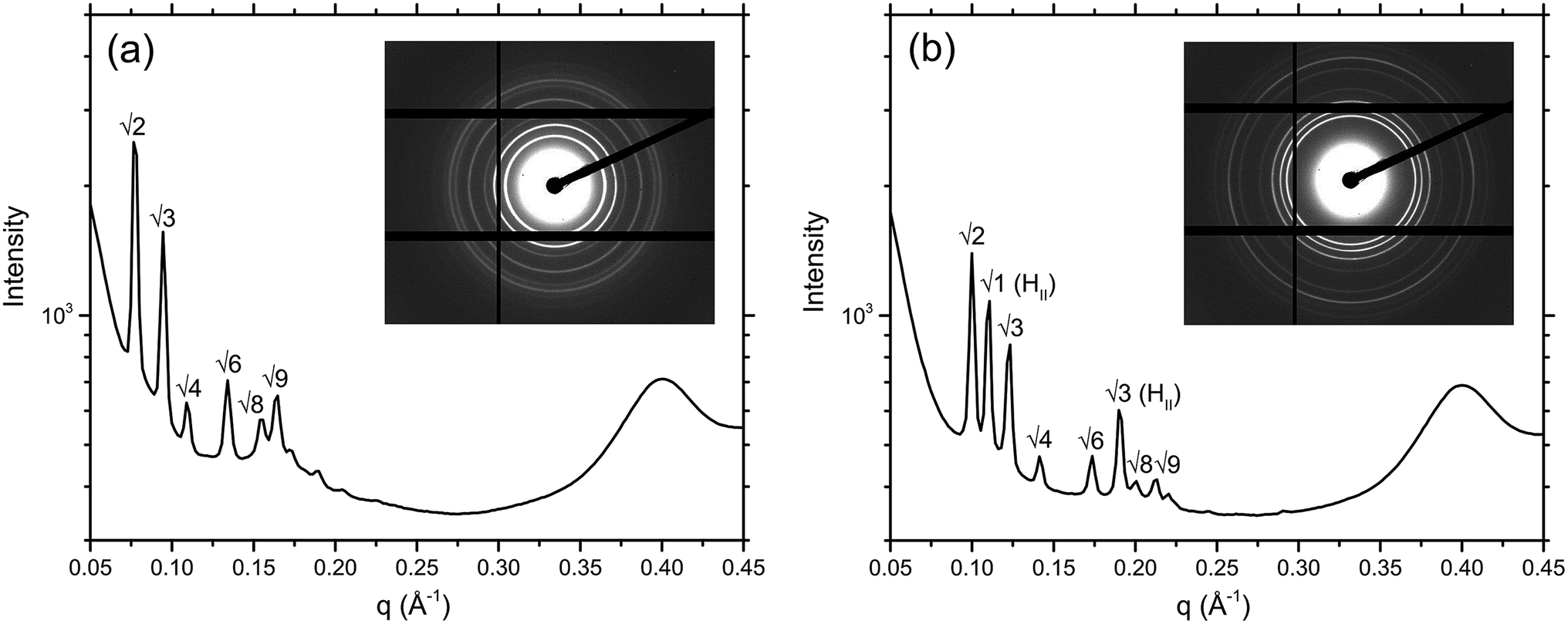 | ||
| Fig. 4 1D diffraction plot of intensity vs. q for: (a) MO + 8 wt% cholesterol illustrating a purely QDII phase, and (b) MO + 8 wt% cholesterol + 2 mol% gA′ illustrating a QDII phase coexisting with an HII phase, with their respective 2D diffraction patterns inset. Also evident is the peak resulting from the plastic of the crystallization plate, seen at 0.4 Å−1. | ||
Circular dichroism
Circular dichroism (CD) spectra were obtained on a Chirascan (Applied Photophysics, Surrey, UK), scanning from 260–200 nm in 1 nm steps, using an equilibration time of 2 s per step. Each spectrum is the average of 3 scans and was smoothed with a 5-point Savitzky–Golay filter. Spectra of cubic phase lipids were recorded in triplicate. The mesophase was prepared by mixing dry peptide into molten lipid at a peptide to lipid ratio of 1:3500, hydrating this mixture at the adequate level to achieve a transparent cubic phase and manually mixing the sample to achieve homogeneity. Each sample was then placed into a two piece 0.1 mm path length quartz cuvette and the spectrum recorded. Spectra in sodium dodecyl sulfate (SDS) were collected in a 1 mm path length quartz cuvette, while those in tetrahydrofuran (THF) were recorded in a 0.1 mm path length cuvette. For mesophase samples, the spectrum of a blank sample of the relevant lipid mesophase without peptide was subtracted from that with incorporated gA′.
Results
Small angle X-ray scattering
The effect of a range of gA′ concentrations (from 0.1 to 15 mol%) on the phase behaviour of the lipidic cubic phases was examined using synchrotron SAXS. Six different lipidic cubic phase systems were studied, varying in both hydrocarbon chain architecture and the structural parameters of the cubic phase (bilayer thickness; water channel diameter). The chemical structure of each of the five lipids used in this work is given in Fig. 5. Data were obtained at 2 h, 12 h, 24 h and 48 h after hydration of the mesophase. Unless otherwise stated all structural trends observed are given at the 2 h time point. The mesophase evolution with time, which was minimal for most samples, is also described.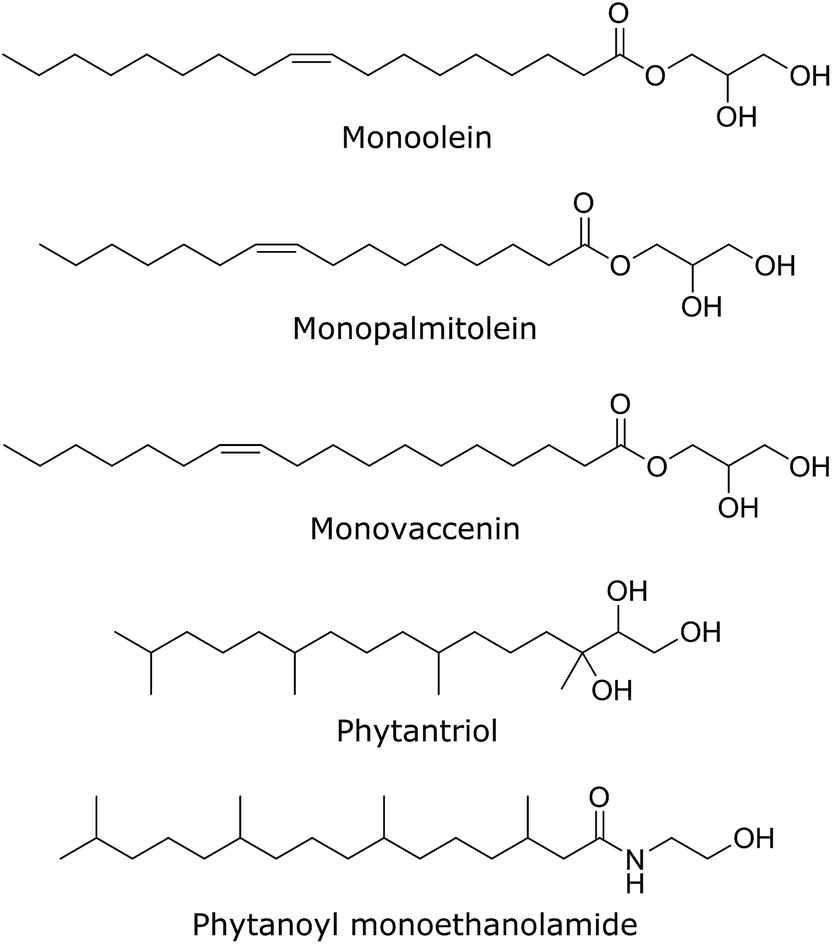 | ||
| Fig. 5 The chemical structures of each of the five bicontinuous cubic phase forming lipids used in our SAXS experiments. | ||
To assess the overall reproducibility of the data, duplicate samples were run at each data-point. The lattice parameters obtained from both data-sets are plotted against each other in the ESI Fig. S1.† The line y = x, representative of an entirely reproducible data-set is provided as a guide to the eye. Note that where coexisting phases were observed, the phase consistent between the two data-points was plotted. The data were highly reproducible for all plates tested, with a difference of 3 Å or less between top and bottom wells in 89% and 96% of wells in 2 and 48 h plates, respectively. Wells that proved irreproducible were discarded from further analysis.
Addition of gramicidin A′ to unsaturated chain lipids
The MAG lipids used (monoolein, monopalmitolein and monovaccenin) all form the diamond QDII bicontinuous cubic phase under the conditions studied (80 wt% H2O, 20 °C, no peptide). The unit cell size was found to increase in the order monoolein (109.8 ± 0.04 Å) < monopalmitolein (115.7 ± 0.2 Å) < monovaccenin (132.1 ± 0.1 Å). These values are in agreement with literature.3The addition of gA′ at increasing concentrations produced similar phase behaviour in all three MAG lipid systems; an initial decrease in lattice parameter with increasing gramicidin concentration was observed with a decrease in the magnitude of the slope approaching 15 mol% peptide. The data points were fitted with a first order exponential decay (Fig. 6), with the fitting parameters listed in the ESI.† For each lipid, the relationship between lattice parameter and gA′ concentration for the Pn3m range was found to be statistically significant (in all cases p < 0.01, with the exception of MO + Chol. at 2 hours, for which p < 0.05). Despite a considerable variation in the initial unit cell size, the lattice parameter at approximately 15 mol% peptide was relatively similar for all three MAGs. For monoolein and monovaccenin, increasing peptide concentration resulted in a phase transition from the QDII phase, to a QDII phase coexisting with an inverse hexagonal (HII) mesophase (at 10 mol% and 3 mol% peptide, respectively). These HII phases coexisted with the QDII phase up to 15 mol% peptide, the highest peptide concentration studied. Additionally, at approximately 15 mol% peptide, both monoolein and monopalmitolein exhibited a coexisting gyroid (QGII) cubic phase with a lattice parameter of 104 and 106.6 Å, respectively. A summary of all phase changes observed from 0 to 15 mol% peptide is provided in Table 1.
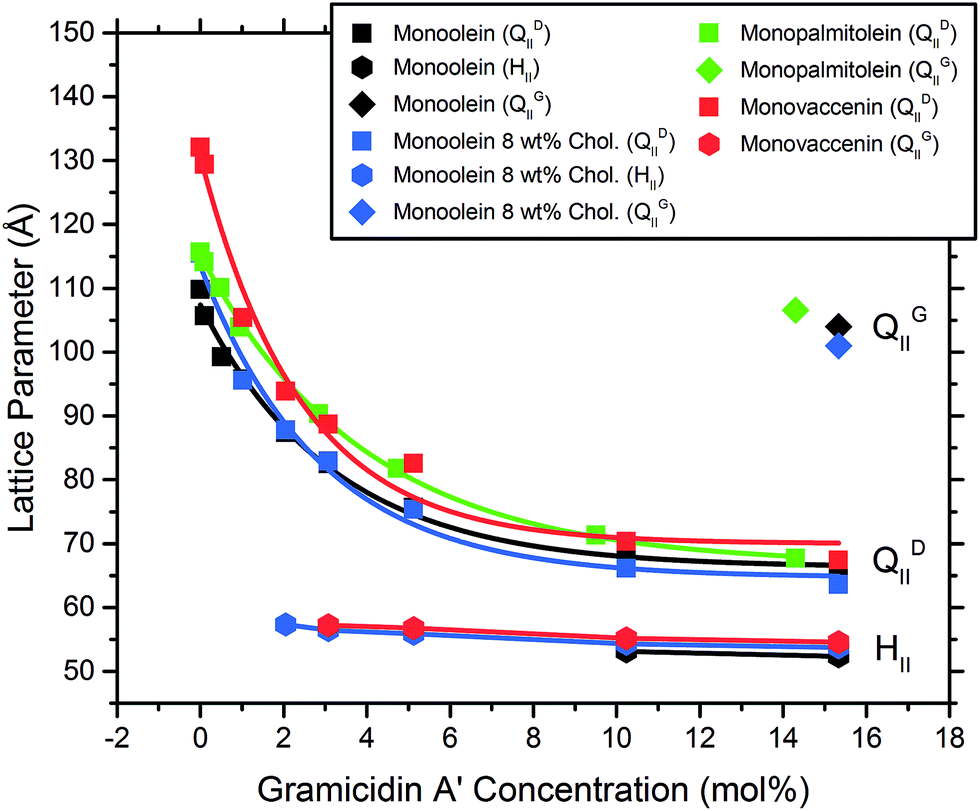 | ||
| Fig. 6 The lattice parameter of MAG lipids as a function of gA′ concentration (mol%) measured at 2 h, showing both the initial QDII cubic phase and the coexisting hexagonal phase occurring at higher peptide concentrations. Values are an average of duplicate samples. | ||
| Lipid | Bilayer thickness (Å) | Observed phase sequence |
|---|---|---|
| Monoolein | 32.1 | QDII → QDII + HII → QDII + HII + QGII |
| Monopalmitolein | 30.3 | QDII → QDII + QGII |
| Monovaccenin | 35.4 | QDII → QDII + HII |
| Monoolein + 8% cholesterol | — | QDII → QDII + HII → QDII + HII + QGII |
| Phytantriol | 29.6 | QDII → QDII + QGII |
| Phytanoyl monoethanolamide | 25.8 | QDII → QDII + HII |
The binary system consisting of monoolein with 8 wt% cholesterol exhibited the QDII → HII transition at lower concentrations of gA′ than pure monoolein. An HII phase coexisting with the QDII phase was observed at 2 mol% gA′ and higher. By 15 mol% gA′, three coexisting phases were observed: QGII, QDII and HII phases, with the QGII lattice parameter measuring 101.0 Å. Whilst the presence of three coexisting phases is not common in pure lipid systems, the Gibbs phase rule allows for up to four coexisting phases for the quaternary system MO–cholesterol–gramicidin A′–H2O at a given temperature and pressure. The change in phase behaviour observed with increasing peptide concentration strongly indicates that gA′ is incorporated within the bilayer. This is consistent with past fluorescence studies and the very low solubility of the peptide in water.25
Addition of gramicidin A′ to branched chain isoprenoid-type lipids
The addition of the same concentrations of gA′ to the isoprenoid-type branched chain lipids phytantriol and phytanoyl monoethanolamide was investigated. Both lipids adopt a QDII mesophase in excess water and in the absence of peptide with a lattice parameter of 69.7 Å and 63.3 Å respectively, significantly smaller than the MAG lipids and in agreement with previous observations.26 The response of the isoprenoid-type cubic phase to addition of gramicidin A′ differed significantly from that of the MAG-type lipids (Fig. 7), where again the data is fitted with a first order exponential decay (fitting parameters are listed in Table S1†). In contrast to the large decrease in lattice parameter observed for the MAG-type lipids, in which the hydrophobic mismatch is greater, addition of gA′ to both phytantriol and phytanoyl monoethanolamide resulted in only a slight decrease in lattice parameter.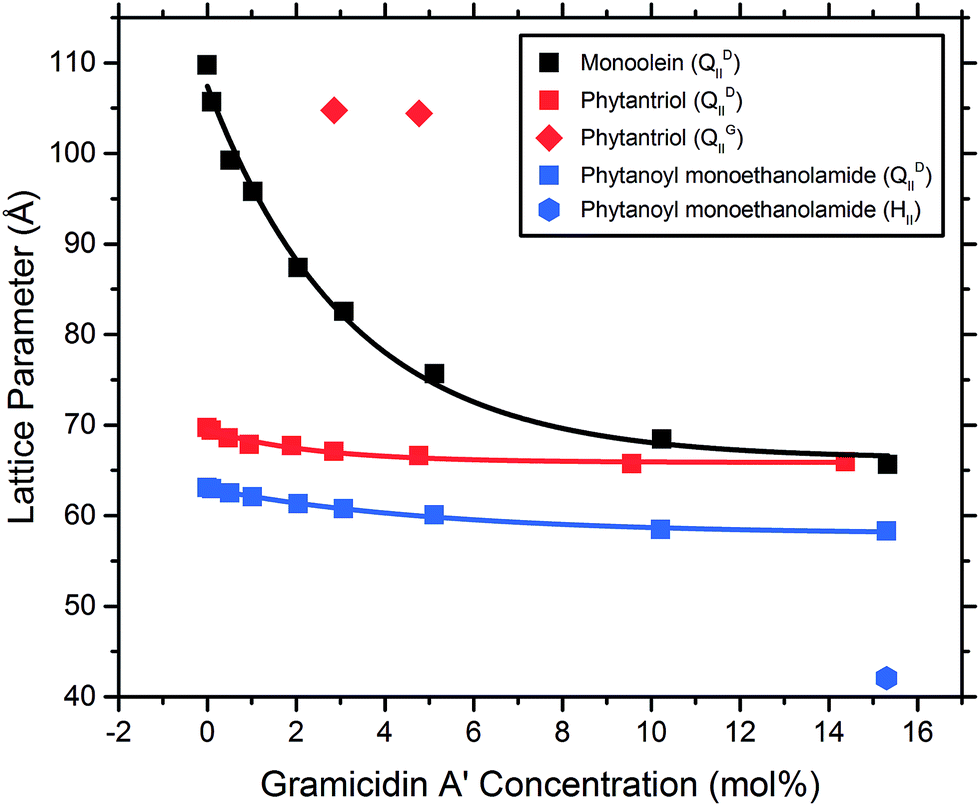 | ||
| Fig. 7 The lattice parameter of bicontinuous cubic phase samples composed of the isoprenoid-type branched chain lipids phytantriol and PE, with monoolein as a representative MAG lipid, plotted as a function of gramicidin A′ concentration (mol%), measured at 2 h. | ||
In phytantriol samples, a coexisting QGII phase with lattice parameter 104.8 and 104.4 Å at 3 and 5 mol% gA′, respectively, was evident. These peaks broadened with increasing peptide concentration such that determination of the lattice parameter was no longer possible, and resulted in a single very wide peak above 10 mol%. The breadth of these peaks is suggestive of a partially ordered system with a gradient of lattice parameters, consistent with a phase undergoing transition. With increasing peptide concentration phytanoyl monoethanolamide displayed the phase sequence QDII → QDII + HII (15 mol%).
Mesophase evolution with time
The mesophase evolution with time over a 48 h period was minimal and in most cases, a given sample retained its phase across the 48 h. However, we note that the timescale for formation of the HII mesophase was slow, and this phase could take up to 48 h to develop, particularly at lower peptide concentrations. For example, for monoolein, the QDII → HII transition occurred at 10 mol% after 2 h equilibration; after 48 h the HII phase was apparent at 5 mol% peptide. The slow kinetics of this transition are especially evident in phytanoyl monoethanolamide samples. Here, at 2 h, the HII phase is apparent only at 15 mol% peptide, coexisting with QDII. At 24 h, PE samples began to show the presence of an HII mesophase at 5 mol% peptide and above, as well as complete disappearance of diffraction peaks corresponding to QDII symmetry at 10 and 15 mol% peptide. At 48 h an HII mesophase at 3 mol% peptide and above was observed, with absence of the cubic mesophase at 5 mol% peptide and higher.Circular dichroism
Bicontinuous cubic phase lipids provide a rigid, transparent and thermodynamically stable matrix to reconstitute peptides in a membrane environment for spectroscopic examination.30 To examine the effects of varying lipid and bilayer parameters on the conformation of gA′, we ran a series of CD experiments. The difficulty associated with producing samples for CD with a path length of less than 0.1 mm limits the range of concentrations at which gA′ can be examined. Furthermore, high levels of peptide result in a less homogeneous cubic phase that varies in excess water point, yielding a sample which is opaque and unsuitable for CD analysis. We, therefore, elected to record CD spectra at a constant peptide to lipid ratio of 1:3500 in each lipid. CD spectra recorded at this peptide to lipid ratio gave a clear signal from the peptide incorporated within the cubic phase.
The two main conformations adopted by gA′, the helical dimer and the double helix, are distinguishable by their characteristic CD spectra. It is believed that the active form in the bilayer is the helical dimer, although this issue is still debated and both conformations may be present in varying ratios.
For comparison with the CD spectra of gA′ in cubic phase lipids, we obtained spectra of the peptide in SDS micelles, where it is predominantly the helical dimer,31,32 and in THF, in which it is present as the double helix conformer.33 In summing these two spectra together, each with a 50% weighting, we obtain a third spectrum which closely resembles that of gramicidin A′ obtained in various lipidic mesophases. We therefore conclude that at the concentrations used in our CD study, gramicidin A′ is present as a mixture of the two conformations, with the magnitude of ellipticity at 220 nm serving as an indication of the ratio of one to the other.
The CD spectra of gA′ encapsulated in MAG lipid bilayers were all very similar (Fig. 8), and indicative of an approximately 50:50 mixture of the helical dimer and the double helix conformations. This result is in agreement with recent observations from Patrick et al., 2016, which demonstrated that the peptide was present as a mixture of conformations under certain conditions.34 Note that for these samples the lipid absorbance caused saturation of the detector and precluded CD analysis below 215 nm. In phytantriol, however, a branched chain lipid, the CD spectra differed from those seen with the MAGS: the ellipticity at 220 nm is well below zero and overall the spectrum more closely resembled that obtained in THF. This suggests that the double helix is present in greater amounts in phytantriol. By fitting a weighted average of the spectra in SDS micelles (representing the helical dimer conformation) and the spectra in THF (representing the double helix) we approximated the contributions of each conformation to the CD spectra observed in the mesophase. For monoolein, the contributions are approximately 39% double helix, 61% helical dimer; and for phytantriol, 62% double helix, 38% helical dimer (see Fig. S2†). Whilst these numbers should not be considered to accurately reflect absolute percentages (the original spectra in THF and SDS micelles are assumed to contain 100% double helix and helical dimer respectively) they do give an indication of the relative amount of each conformation present. These results confirm the influence of lipid bilayer properties on the adopted conformation and will be discussed further in the following section.
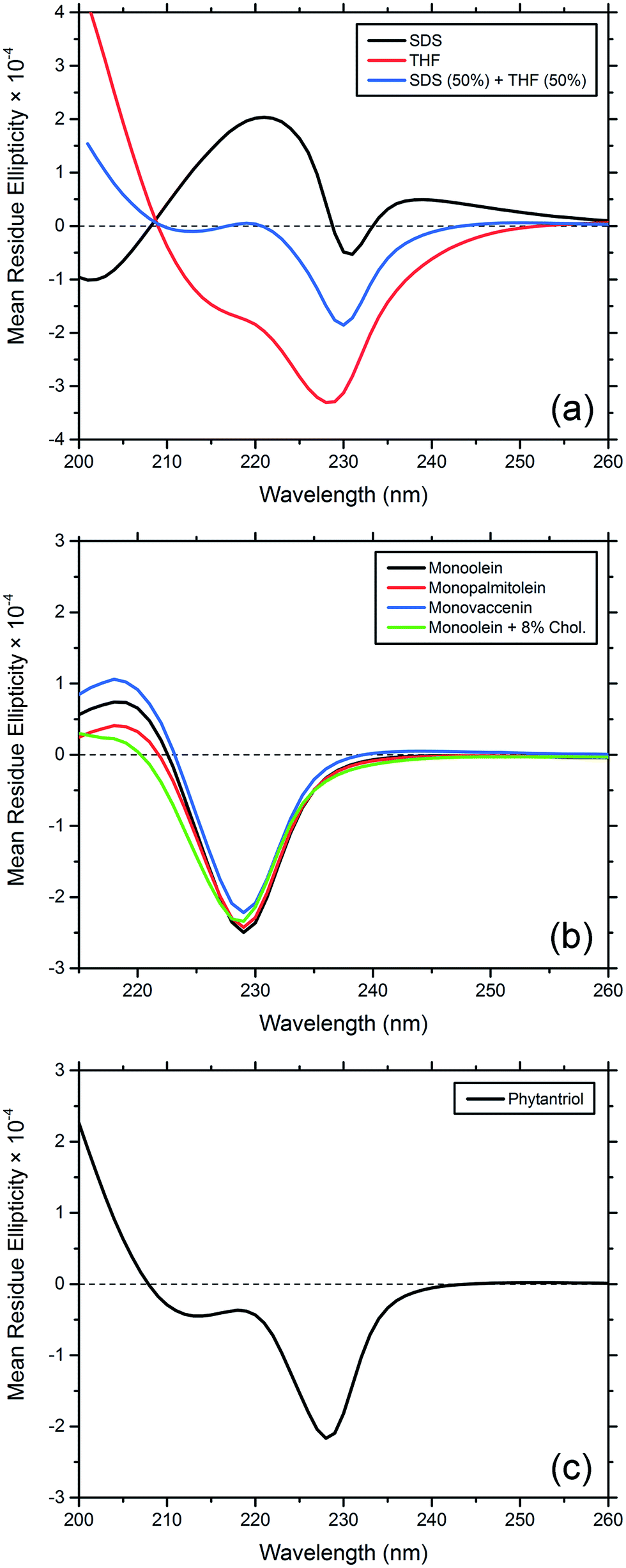 | ||
| Fig. 8 Circular dichroism spectra of gramicidin A′ encapsulated in bicontinuous cubic phases comprised of various lipids. (a) CD spectra of gA′ in THF, encapsulated in SDS micelles and the result of summing these two spectra each with a 50% weighting. (b) CD spectra of gA′ encapsulated in monoolein, monopalmitolein, monovaccenin and monoolein + 8 wt% cholesterol. (c) CD spectrum of gA′ encapsulated in phytantriol cubic phase. | ||
Discussion
The complex structural relationship between proteins and peptides and the lipid mesophase is influenced by a number of interconnected factors. There is a large body of evidence suggesting that hydrophobic mismatch, the difference between the length of the membrane spanning region of a guest molecule and the hydrophobic region of the bilayer, is crucial in determining the functionality of embedded peptides and proteins.35 It has also been demonstrated that the degree of mismatch influences bilayer behaviour, with the bilayer altering both its thickness and the overall mesophase structure to accommodate the hydrophobic region of peptides and proteins. The lateral pressure profile of a bilayer system is also highly influential, with changes in local lateral pressure influencing not just the behaviour of the bilayer but also the conformations of embedded molecules.36 The spontaneous curvature of these non-lamellar forming lipids is linked to the lateral pressure profile and must also be considered, although these parameters can be difficult to deconvolute. Studies with bacteriorhodopsin have demonstrated that the spontaneous curvature of a bilayer system is important in determining both the location and tertiary structure of biomolecules embedded in the bilayer.37,38The differences in mesophase behaviour of each of the different MAG lipids, may reflect differences in bilayer thickness and hence hydrophobic mismatch with the peptide. From the lattice parameters measured at 0 mol% peptide we can estimate the bilayer thickness via the infinitely periodic minimal surface model (IPMS) outlined by Turner et al.39Eqn (1) gives the lipid fraction (Φl) for a sample of known composition:
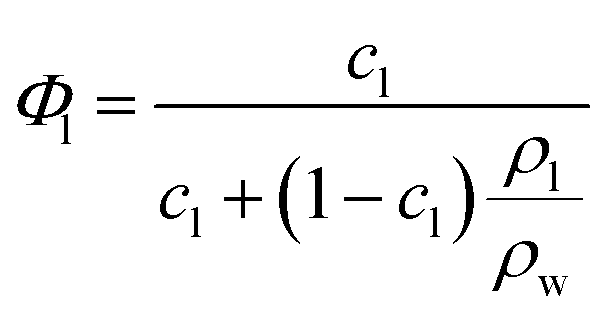 | (1) |
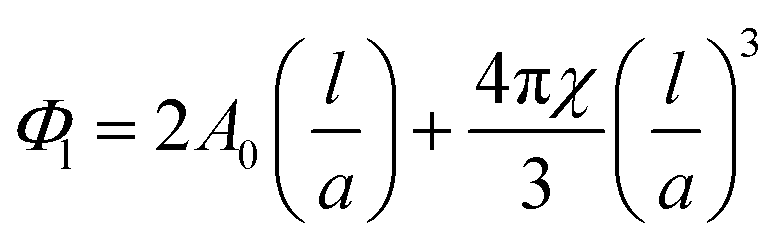 | (2) |
Given the lipid fraction, and the lattice parameter in Ångstroms (a) eqn (2) can be solved for lipid length (l). Here A0 is the dimensionless surface area, given by A0 = A/V2/3, where A is the surface area per unit cell of volume V = a3. The Euler characteristic χ provides a measure of the integrated Gaussian curvature per unit area of the surface. These values are specific to each cubic phase symmetry (A0 = 1.919 and χ = −2 for QDII). The lipid length is doubled to give the bilayer thickness values shown in Table 1, increasing in the order PE < PT < MP < MO < MV (this was not calculated for the cholesterol containing samples as the excess water point of this mixture is unknown).
A comparison of the channel length of the helical dimer gA′ conformation (believed to be the active form present within the bilayer, and the conformation responsible for HII formation40) with the bilayer thickness of each cubic phase MAG lipid suggests that hydrophobic mismatch (defined as the lipid length minus the peptide length) plays a significant role in determining lipid phase behaviour. The length of the helical dimer conformation is 25 Å, so that the hydrophobic mismatch for monovaccenin (−10.4 Å) is significantly larger than for monopalmitolein (−5.3 Å).
We find that the concentration at which the HII phase is formed is correlated with bilayer thickness and hence hydrophobic mismatch. The hydrophobic mismatch between the gA′ and the bilayer is greatest for MV, which has a calculated bilayer thickness of 35.4 Å in excess water. An HII phase is observed at relatively low peptide concentrations (3 mol% gA′) for this system. As the hydrophobic mismatch is reduced the peptide concentration at which the HII phase is observed increases. Thus, for MO, with a bilayer of 32.1 Å, an HII phase is not observed until 10 mol% gA′. For MP, which has the thinnest bilayer of the MAGs (30.3 Å) and hence a smaller hydrophobic mismatch, we note the absence of the HII phase even at the highest concentrations of peptide. Our results are in agreement with previous research indicating that greater hydrophobic mismatch leads to the formation of the HII phase at lower concentrations of gA′.25 The length of the gramicidin dimer appears to be a driver of phase behaviour for all lipids. This is supported by the observation that, despite a wide variation in the initial lattice parameter, the lattice parameters of all cubic phases tested approach a similar value at high peptide concentrations (Fig. 6 and 7). We suggest that peptide incorporation causes a thinning of the bilayer and concomitant reduction in lattice parameter, and that for all lipids this reduction continues until the cubic phase bilayer thickness matches the hydrophobic span of the peptide.
The ability of cholesterol to induce a coexisting HII phase at lower peptide concentrations in monoolein may be explained by its ‘rigidification’ or ordering effect on the bilayer.41 This effect has been observed in other studies42 at concentrations similar to those used here. We suggest that the increase in bilayer rigidity (or bending modulus) associated with the addition of cholesterol reduces the ability of the QDII phase to accommodate the gA′ by increasing its intrinsic curvature; triggering a phase transition to HII at lower concentrations of peptide.
The effects of variations in hydrophobic mismatch between peptides and membranes have been previously explored by Killian et al.43 on WALP peptides (peptides consisting of a repeating Leu–Ala chain capped on either end with Trp, designed to model transmembrane peptides) in diacylphosphatidylcholines of various lengths. The resultant phase of these systems is highly dependent on hydrophobic mismatch. Systems with a negative mismatch preferentially form HII phases, while small positive mismatches formed isotropic phases, and large positive mismatches formed bilayer structures. Similar behaviour was observed for gA′.44
Gramicidin A′ and similar peptides are proposed to promote HII formation by increasing the volume of the hydrocarbon region,31,45 and then occupying the positions in the HII phase in which the monolayer thickness is smallest.40 In the absence of peptide, the HII phase is generally not observed until much higher temperatures, e.g. around 90 °C for hydrated monoolein,46 due to the prohibitive steric strain imposed by the high curvature of the HII phase. These explanations conform well to our observations that at high peptide concentrations the resultant HII phases of MAG lipids all possess similar lattice parameters.
For monoolein, monoolein + 8 wt% cholesterol, monopalmitolein, and phytantriol samples, a QGII cubic was seen to coexist with a HII and/or a QDII cubic phase at 14–15 mol% peptide. This coexistence is unlikely to reflect a thermodynamically stable state, and likely results from the mixture of gramicidin A′ conformations present. The QDII → QGII transition was confirmed through the use of the Bonnet ratio: the predicted ratio between lattice parameters of coexisting cubic mesophases in excess water under equilibrium conditions.47 For example, in phytantriol with 3 mol% gA′ we observed both QDII (lattice parameter of 67.1 Å) and QGII (lattice parameter of 104.8 Å), the ratio of which (1.562) closely matches with that of the Bonnet ratio for coexisting QDII/QGII mesophases (1.576).
However, in comparing the disparate phase behaviour of monopalmitolein and phytantriol, two lipids with relatively similar bilayer thickness, it becomes clear that the bilayer thickness is not the only factor governing phase behaviour. Here we must also consider differences in the lateral pressure profile between the different lipid bilayers. For the MAG lipids, similarities in both the headgroup and the acyl chains will result in a very similar lateral pressure profile for these three lipids. However, previous work based on MD simulations has indicated that the local lateral pressure exerted within branched chain lipids is much higher than within unsaturated lipid chains. This has been demonstrated by Miklavcic et al. in their molecular dynamics study of branched chain archaeal lipid bilayers.27
To probe the effect of variations in lipid structure on the peptide conformation, CD spectra were obtained for gA′ in the three MAG lipids, MO + 8 wt% cholesterol, phytantriol, THF and SDS micelles. The spectra obtained for MO, MP, MV and MO + 8 wt% cholesterol are similar. For these unsaturated cubic phase lipids, the CD spectra resembles neither the helical dimer conformation in SDS micelles nor the double helix observed in THF, but appears to reflect a mixture of the two, with the proportion of the helical dimer conformation increasing in the order MO + 8 wt% cholesterol < MP < MO < MV. The proportion of the helical dimer conformation increases with bilayer thickness, while the reduction in helical dimer conformation caused by the addition of cholesterol is likely due to increased bilayer pressure, a known effect of cholesterol on the bilayer.48 The CD spectrum measured in phytantriol similarly represents a mixture of the two conformations. However, the drop in the ellipticity at 220 nm brings the spectrum closer to that found in THF solution, and hence is consistent with a larger proportion of the double helix conformation. An examination of the channel widths reveals that the double helix (β5.6) conformation is narrower (0.95 Å maximum pore radius) than the helical dimer (β6.3, 1.80 Å maximum pore radius).49
It should be noted that in our CD samples gramicidin A′ was added at a peptide to lipid ratio of 1:3500, and at higher concentrations, such as those used in our SAXS study, peptide–peptide interactions may play a larger role in determining secondary structure. Even so, these results show that bilayer properties are capable of altering peptide conformation, and are consistent with the theory that peptides and proteins adopt narrower conformations in response to increased local lateral pressure within the bilayer.36
The finding that the branched chain lipid phytantriol stabilises the double helix conformation agrees well with our observations of cubic phase behaviour. The double helix is significantly longer (35 Å) than the helical dimer (25 Å),49 and as such should not promote an HII phase, but rather an isotropic or bilayer structure. This is consistent with our observations that branched chain lipids exhibit small changes in phase behaviour with gramicidin A′ addition compared to MAGs.
In examining the dissimilar phase behaviour of the MAG and isoprenoid type lipids subsequent to gA′ addition, we must also look at the intrinsic curvature of the bilayer systems involved. PT and PE, with lattice parameters much smaller than the MAG lipids tested, have an increased intrinsic curvature. It should be noted that although the total mean curvature of the surface is zero, the Gaussian curvature is everywhere non-positive and varies between zero at the flat points and most negative at the saddle points. With our CD experiments we observed that in PT, the double helix conformation is present in greater amounts than in the MAGs. Previous work by Kulkarni et al. has shown that the integral membrane protein bacteriorhodopsin may preferentially select a region of the bilayer based on Gaussian curvature, and will necessarily alter its tertiary structure to accommodate the principal curvature at these points.
The spontaneous curvature of lipid systems has also been shown to influence the uptake of hydrophobic antimicrobial peptides. Using EPR spectroscopy Cafiso et al. found that the binding of alamethicin to membranes consisting of DOPC/DOPE decreased with increasing spontaneous curvature, which was accomplished by increasing the DOPE content.50 The increase in binding free energy was found to be linearly dependent on the increase in DOPE mol fraction. This effect may be responsible for the exponential decay observed in our SAXS results; as curvature increases with increasing gA′ concentration, the ability of the membrane to bind further peptide is reduced. Furthermore, the decreased effect of gA′ on PT and PE lipid membranes may also stem from decreased uptake, an effect of the increased spontaneous curvature in these lipid systems.
Conclusions
The MAG host lipids responded to increasing concentrations of gramicidin A′ with a sharp decrease in unit cell size, and either retained cubic QDII symmetry or transitioned to an HII mesophase at high peptide concentrations, an effect that was correlated with bilayer thickness. A second class of lipids featuring isoprenoid-type branched chains, reacted differently and transitioned to either an HII phase or a cubic QGII phase with very little change in QDII lattice parameter. These observations support the view that hydrophobic mismatch between bilayer structures and guest molecules is an important factor in determining the nature of subsequent changes in lipid mesophase structure. The spontaneous curvature and lateral pressure profile of the lipid bilayer may also play a central role, evidenced by the significant differences in phase behaviour displayed by the branched chain lipids. Additionally, this work shows that phase behaviour trends observed subsequent to peptide addition in non-cubic lipidic structures (such as the work by Killian et al.) can serve as good predictors of cubic system phase behaviour.Through the use of circular dichroism we have shown that peptides are able to adopt different conformations depending on the lipid mesophase environment. We suggest that this is due to differences in the lateral pressure profiles of these two lipid classes. Increasing the local lateral pressure within the bilayer may result in the peptide adopting a narrower conformation, consistent with previous observations.34
These results should be of use in the planning of in meso crystallization trials, where careful lipid selection is needed to retain desired protein conformations, as well as the design of peptide and protein based drug delivery and biosensor systems incorporating bicontinuous cubic phase materials.
Acknowledgements
The authors acknowledge the use of the SAXS/WAXS beamline at the Australian Synchrotron. T.G.M. was the recipient of a CSIRO-Melbourne Research Scholarship. C.E.C. is the recipient of an ARC DECRA Fellowship DE160101281. We wish to thank Dr Shane Seabrook and Dr Janet Newman at the C3 Collaborative Crystallization Centre, CSIRO, Melbourne, Australia.References
- C. E. Conn and C. J. Drummond, Soft Matter, 2013, 9, 3449–3464 RSC.
- M. Caffrey and V. Cherezov, Nat. Protoc., 2009, 4, 706–731 CrossRef CAS PubMed.
- C. Fong, T. Le and C. J. Drummond, Chem. Soc. Rev., 2012, 41, 1297–1322 RSC.
- X. Mulet, C. E. Conn, C. Fong, D. F. Kennedy, M. Moghaddam and C. J. Drummond, Acc. Chem. Res., 2013, 46, 1497–1505 CrossRef CAS PubMed.
- X. Mulet, B. J. Boyd and C. J. Drummond, J. Colloid Interface Sci., 2012, 393, 1–20 CrossRef PubMed.
- V. Luzzati, A. Tardieu, T. Gulikkrz, E. Rivas and F. Reisshus, Nature, 1968, 220, 485–488 CrossRef CAS PubMed.
- A. Taha, http://www.k3dsurf.sourceforge.net/, 2007.
- http://www.blender.org/, 2015.
- J. C. Shah, Y. Sadhale and D. M. Chilukuri, Adv. Drug Delivery Rev., 2001, 47, 229–250 CrossRef CAS PubMed.
- B. J. Boyd, in Bicontinuous liquid crystals, ed. M. L. Lynch and P. T. Spicer, 2005, pp. 285–306 Search PubMed.
- B. Ericsson, P. O. Eriksson, J. E. Lofroth and S. Engstrom, ACS Symp. Ser., 1991, 469, 251–265 CrossRef CAS.
- A. Zabara, R. Negrini, O. Onaca-Fischer and R. Mezzenga, Small, 2013, 9, 3602–3609 CrossRef CAS PubMed.
- C. E. Conn, C. Darmanin, S. M. Sagnella, X. Mulet, T. L. Greaves, J. N. Varghese and C. J. Drummond, Soft Matter, 2010, 6, 4828–4837 RSC.
- D. D. Busath, Annu. Rev. Physiol., 1993, 55, 473–501 CrossRef CAS PubMed.
- B. A. Wallace, J. Struct. Biol., 1998, 121, 123–141 CrossRef CAS PubMed.
- D. A. Kelkar and A. Chattopadhyay, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 2011–2025 CrossRef CAS PubMed.
- N. Hofer, D. Aragao and M. Caffrey, Biophys. J., 2010, 99, L23–L25 CrossRef PubMed.
- S. Liu, M. Yu, Y. He, L. Xiao, F. Wang, C. Song, S. Sun, C. Ling and Z. Xu, Hepatology, 2008, 47, 1964–1973 CrossRef CAS PubMed.
- H. A. Lashuel, D. Hartley, B. M. Petre, T. Walz and P. T. Lansbury, Nature, 2002, 418, 291 CrossRef CAS PubMed.
- S. Bhakdi and J. Tranumjensen, Microbiol. Rev., 1991, 55, 733–751 CAS.
- R. Sarges and B. Witkop, J. Am. Chem. Soc., 1964, 86, 1862–1863 CrossRef CAS.
- B. A. Wallace, Annu. Rev. Biophys. Biophys. Chem., 1990, 19, 127–157 CrossRef CAS PubMed.
- V. F. Bystrov and A. S. Arseniev, Tetrahedron, 1988, 44, 925–940 CrossRef CAS.
- G. A. Woolley and B. A. Wallace, J. Membr. Biol., 1992, 129, 109–136 CAS.
- W. Liu and M. Caffrey, J. Struct. Biol., 2005, 150, 23–40 CrossRef CAS PubMed.
- S. M. Sagnella, C. E. Conn, I. Krodkiewska and C. J. Drummond, Soft Matter, 2009, 5, 4823–4834 RSC.
- A. Polak, M. Tarek, M. Tomsic, J. Valant, N. P. Ulrih, A. Jamnik, P. Kramar and D. Miklavcic, Langmuir, 2014, 30, 8308–8315 CrossRef CAS PubMed.
- L. van't Hag, C. Darmanin, T. C. Le, S. Mudie, C. E. Conn and C. J. Drummond, Cryst. Growth Des., 2014, 14, 1771–1781 Search PubMed.
- J. M. Seddon, A. M. Squires, C. E. Conn, O. Ces, A. J. Heron, X. Mulet, G. C. Shearman and R. H. Templer, Philos. Trans. R. Soc., A, 2006, 364, 2635–2655 CrossRef CAS PubMed.
- E. M. Landau and P. L. Luisi, J. Am. Chem. Soc., 1993, 115, 2102–2106 CrossRef CAS.
- B. A. Cornell, F. Separovic, A. J. Baldassi and R. Smith, Biophys. J., 1988, 53, 67–76 CrossRef CAS PubMed.
- A. L. Lomize, V. Orekhov and A. S. Arsen'ev, Bioorg. Khim., 1992, 18, 182–200 CAS.
- M. C. Bano, L. Braco and C. Abad, FEBS Lett., 1989, 250, 67–71 CrossRef CAS PubMed.
- J. W. Patrick, R. C. Gamez and D. H. Russell, Biophys. J., 2016, 110, 1826–1835 CrossRef CAS PubMed.
- D. A. Kelkar and A. Chattopadhyay, Biochim. Biophys. Acta, Gen. Subj., 2007, 1768, 1103–1113 CrossRef CAS PubMed.
- R. S. Cantor, J. Phys. Chem. B, 1997, 101, 1723–1725 CrossRef CAS.
- M. Grabe, J. Neu, G. Oster and P. Nollert, Biophys. J., 2003, 84, 854–868 CrossRef CAS PubMed.
- C. V. Kulkarni, A. M. Seddon, O. Ces and R. H. Templer, Soft Matter, 2010, 6, 4339–4341 RSC.
- D. C. Turner, Z. G. Wang, S. M. Gruner, D. A. Mannock and R. N. McElhaney, J. Phys. II, 1992, 2, 2039–2063 CrossRef CAS.
- J. A. Killian, Biochim. Biophys. Acta, Gen. Subj., 1992, 1113, 391–425 CrossRef CAS.
- M. Bloom, E. Evans and O. G. Mouritsen, Q. Rev. Biophys., 1991, 24, 293–397 CrossRef CAS PubMed.
- J. Lemmich, K. Mortensen, J. H. Ipsen, T. Honger, R. Bauer and O. G. Mouritsen, Eur. Biophys. J. Biophys. Lett., 1997, 25, 293–304 CrossRef CAS PubMed.
- J. A. Killian, I. Salemink, M. R. de Planque, G. Lindblom, R. E. Koeppe 2nd and D. V. Greathouse, Biochemistry, 1996, 35, 1037–1045 CrossRef CAS PubMed.
- B. A. Cornell and F. Separovic, Eur. Biophys. J., 1988, 16, 299–306 CAS.
- B. A. Cornell, L. E. Weir and F. Separovic, Eur. Biophys. J. Biophys. Lett., 1988, 16, 113–119 CAS.
- J. Briggs, H. Chung and M. Caffrey, J. Phys. II, 1996, 6, 723–751 CrossRef CAS.
- S. Andersson, S. T. Hyde, K. Larsson and S. Lidin, Chem. Rev., 1988, 88, 221–242 CrossRef CAS.
- M. Patra, Eur. Biophys. J. Biophys. Lett., 2005, 35, 79–88 CrossRef CAS PubMed.
- O. S. Smart, J. M. Goodfellow and B. A. Wallace, Biophys. J., 1993, 65, 2455–2460 CrossRef CAS PubMed.
- J. R. Lewis and D. S. Cafiso, Biochemistry, 1999, 38, 5932–5938 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Plots of lattice parameters for top vs. bottom well (Fig. S1); the fitting parameters resulting from fitting a first order decay function to the plots of lattice parameter (Å) vs. gA′ concentration (mol%) (Table S1); comparison of the CD spectra of gA′ encapsulated in monoolein and phytantriol with a weighted, intensity adjusted combination of the spectra of gA′ in SDS micelles and THF (Fig. S2). See DOI: 10.1039/c6ra13658c |
| This journal is © The Royal Society of Chemistry 2016 |