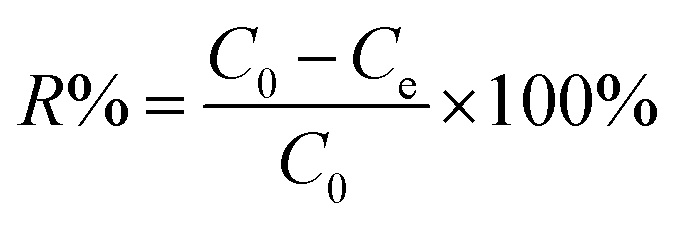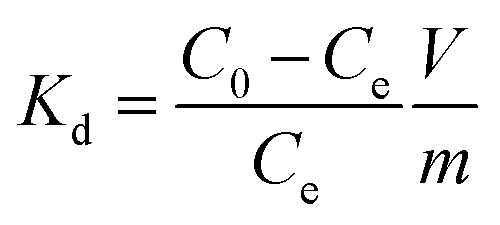DOI:
10.1039/C6RA14193E
(Paper)
RSC Adv., 2016,
6, 69947-69955
Mesoporous hydroxylapatite/activated carbon bead-on-string nanofibers and their sorption towards Co(II)†
Received
1st June 2016
, Accepted 17th July 2016
First published on 18th July 2016
Abstract
The present work aims to prepare mesoporous hydroxylapatite/activated carbon (meso-HA/AC) bead-on-string nanofibers and evaluate their sorption towards Co(II) via sorption kinetics and isotherms. Using polyvinyl alcohol/hydroxylapatite/glucose electrospun nanofibers as the precursor, meso-HA/AC nanofibers are prepared by a hydrothermal process. The nanofibers show bead-on-string structures with mesoporous characteristics, main pores around 29 nm in size, and a specific surface area of 30.014 m2 g−1. Moreover, the rod-like HA crystals assemble into bundles and insert into the two neighboring activated carbon beads. The sorption of Co(II) onto meso-HA/AC nanofibers is strongly dependent on pH and ionic strength. The pseudo-second-order model is valid to describe the sorption of Co(II) onto meso-HA/AC nanofibers, and the intraparticle diffusion is not the sole rate-controlling step. Both Langmuir and Freundlich models can well describe the sorption isotherms, and the Langmuir model is slightly better than the Freundlich model. Moreover, the thermodynamic parameters imply that the sorption process is spontaneous and endothermic. The meso-HA/AC bead-on-string nanofibers may have potential as a highly effective material for the removal of heavy metal ions from aqueous solution.
1. Introduction
Heavy metal ion pollution has become a serious environmental problem and the removal of heavy metal ions from water resources has attracted considerable attention.1–3 Cobalt(II) is a very toxic element affecting the environment, and living creatures, especially, radionuclide 60Co(II) is considered to be one of the most dangerous radionuclides because of its relatively long half live (T1/2 = 5.27 a).4 A high dose Co(II) may lead to some health problems such as diarrhea, lung irritation, bone defects, paralysis and low blood pressure.1,5–7 The permissible limits of cobalt in irrigation water and livestock wastewater are 0.05 and 1.0 ppm, respectively.8,9 Therefore, the removal of Co(II) from aqueous solution is significant for environmental protection and public health.
Various methods are available for removal of heavy metals including chemical precipitation,10 electrodialysis,11 reverse osmosis,12 ion exchange,13 membrane process,14 solvent extraction,15 and sorption.16–19 Among all these methods, sorption is considered as one of the most effective alternatives and widely used method due to its high efficiency, moderate operational conditions, economical feasibility and availability of different adsorbents.2,20 Many achievements have been acquired on cobalt(II) removal from aqueous solutions through sorption by using minerals or its composites: kaolinite,21,22 bentonite,23 hectorite,24 hydroxyapatite,25,26 and chitosan-coated perlite beads27 etc. Herein, hydroxyapatite (HA, Ca10(PO4)6(OH)2), a bioactive and biocompatible ceramic, is one of the most promising materials for removal of different toxic elements because of its easy availability, low cost, high capacity, and moderate solubility.25,28–30
Recently, porous nanofibers have caused considerable concern in heavy metals from aqueous solutions due to their relatively large surface area, small inter-fibrous pore size, and well-modified surface properties.31–33 It is well known that electrospinning technique is an efficient method for fabrication of sub-micron or nanoscale fibers. Using electrospun composite fibers as precursors, it is confirmed to be an effective and convenient method to fabricate porous inorganic nanofibers by calcination process (e.g., mesoporous titanium dioxide fibers,34 mesoporous hydroxylapatite/zinc oxide nanofibers,35 LaFeO3 and LaNiO3 porous hollow nanofibers36,37). Additionally, hydrothermal method is also a general and widely used technique to fabricate porous nanoparticles (e.g., mesoporous TiO2,38 porous MgO microrods,39 and mesoporous silica materials40) for removal of heavy metals from the aqueous solution. Encouraged by these achievements, the hydrothermal method is developed to fabricate mesoporous nanofibrous absorbent using electrospun composite fibers as precursors in the present work.
We aimed to fabricate hydroxylapatite/activated carbon (meso-HA/AC) bead-on-string nanofibers from polyvinyl alcohol/hydroxylapatite/glucose (PVA/HA/glucose) electrospun nanofibers via hydrothermal process, evaluate the removal efficiency of the nanofibers toward Co(II) by sorption kinetics and sorption isotherms, and investigate the sorption behaviors of Co(II) on meso-HA/AC nanofibers as a function of pH, ionic strength, and thermodynamic parameters. Additionally, the structure and morphology of the nanofibers were studied.
2. Material and methods
2.1 Materials
Polyvinyl alcohol (PVA, polymerization degree 1750 ± 50, alcoholysis degree 98%), ammonium dihydrogen phosphate (NH4H2PO4), calcium nitrate tetrahydrate (Ca(NO3)2·4H2O), ammonium hydroxide solution (NH3·H2O, 25–28%) and glucose (D-glucose monohydrate, 198 g mol−1) were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). All the chemical reagents used were of analytical grade.
2.2 Preparation of samples
2.2.1 PVA/HA/glucose nanofibers. A PVA aqueous solution (9.3%, w/w) was prepared by dissolving 4.2 g PVA in 45 g distilled water at 95 °C for 4 h under stirring. According to our experimental results, Ca(NO3)2·4H2O (2.0 g) was added into above PVA solution and the pH value of the solution was adjusted to 9.0 by NH3·H2O, and then a stoichiometric amount of NH4H2PO4 solution (2.0 mol L−1) was added dropwise into the solution within an hour at room temperature assisted with stirring and kept the stirring for an additional 1 h. Finally, the needed glucose (HA/glucose ratios were at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:3, 1:5 and 1:10, w/w, respectively) was dissolved in distilled water (5.0 g), and then added to the above solution. After being stirred for 1 h, the corresponding PVA/HA/glucose electrospinning solutions with diffident HA/glucose ratios were obtained.
:1, 1:3, 1:5 and 1:10, w/w, respectively) was dissolved in distilled water (5.0 g), and then added to the above solution. After being stirred for 1 h, the corresponding PVA/HA/glucose electrospinning solutions with diffident HA/glucose ratios were obtained.The PVA/HA/glucose nanofibers were fabricated on an electrospinning device consisting of a syringe, a needle (0.41 mm internal diameter), a copper sheet, a ground electrode and a high-voltage power supply (DW-P403-1ACCC, Tianjin Dongwen, China). On the basis of experimental data, electrospinning processing was carried out at room temperature with relative humidity (RH) at 50%. The supplied voltage and tip-to-collector distance (TCD) were kept at 22 kV and 15 cm, respectively. The nanofibers were dried at 50 °C under vacuum condition for 24 h.
2.2.2 Mesoporous hydroxylapatite/activated carbon composite nanofibers. All hydrothermal reactions were conducted in the same manner. The collected PVA/HA/glucose dry nanofibers were putted into a 50 mL Teflon autoclave liner with 35 mL distilled water. The liner was then sealed in a stainless steel autoclave and placed into a box furnace for the desired three temperatures (160, 180, and 200 °C) and four hours (12, 16, 24, and 48 h). At the end of reaction, the autoclave was slowly cooled down to room temperature. After washed twice with distilled water and absolute ethanol in turn, the resulting mesoporous hydroxylapatite/activated carbon (meso-HA/AC) composite nanofibers were dried in a vacuum oven at 50 °C for 24 h.
2.3 Morphology and structure
The morphologies of nanofibers were observed using scanning electron microscopy (SEM, SU8020, Hitachi, Japan) under an accelerating voltage of 5 kV, and all of the specimens were sputter-coated with a layer of gold. SEM images were analyzed by Image Tool software, and a total of 50 counts were used to calculate the average diameter of nanofibers. Transmission electron microscopy (TEM, H-7650, Hitachi, Japan) images were recorded at an acceleration voltage of 160 kV. The sample was ultrasonically treated with absolute ethanol at room temperature for 3 h and then a highly diluted mixture was dropped on 200-mesh copper grids coated with carbon. Nitrogen adsorption–desorption experiment was performed at 77 K using a Beckman Coulter SA 3100 (Beckman Coulter, CA, USA).
Fourier transformed infrared spectroscopy (FTIR) spectra were measured on a Nicolet 6700 spectrometer (Thermo Nicolet, Madison, WI, USA) in the range of 4000–400 cm−1 using KBr pellets. Raman spectra were recorded using a confocal micro-Raman spectrometer (LabRAM HR-800, Horiba Jobin Yvon, France) with a He–Ne laser excitation at 633 nm. X-ray diffraction (XRD) patterns were obtained using an X-ray diffractometer (X'Pert Pro MPD, Philips, Netherlands) with Cu Kα radiation (λ = 0.15406 nm) and operated at 40 kV and 40 mA.
2.4 Sorption experimental procedures
The sorption experiments of Co(II) onto nanofibers were carried out by batch technique in polyethylene centrifuge tubes under ambient conditions. The stock suspensions of meso-HA/AC nanofibers and NaNO3 solution were pre-equilibrated for 12 h, and then Co(II) stock solution was added to achieve the desired concentrations of different components. The pH values of the suspensions were adjusted to desired values by adding negligible volumes of 0.01 or 0.1 mol L−1 HNO3 or NaOH solutions. On the basis of our experimental results (Fig. S1, ESI†), a concentration of meso-HA/AC nanofibers of 0.8 g L−1 was used in the sorption experiments as an optimum value. For the sorption experiment, the above solutions were placed on a shaker platform (120 rpm) for 24 h to assure the sorption equilibrium, and then centrifuged at 8000 rpm for 25 min. The Co(II) concentration within supernatant was analyzed using a UV-754 PC spectrophotometer (Shanghai Jinghua, China) and calculated according to the linear regression equation from the calibration curve (Fig. S2, ESI†). The sorption percentage (R%) and distribution coefficient (Kd) were calculated as follows:| |
 | (1) |
| |
 | (2) |
where C0 (mol L−1) is the initial Co(II) concentration, Ce (mol L−1) is the residual Co(II) concentration at sorption equilibrium, m (g) is the mass of sample, and V (L) is the volume of suspension.
2.5 Statistical analysis
All experiments were performed in triplicate. Statistical analysis was analyzed using the unpaired Student's t-test (n = 3), and the values were expressed as the means ± standard deviation (SD). The threshold for statistical significance was set at p < 0.05.
3. Results and discussion
3.1 Effects of HA/glucose ratio on morphology of PVA/HA/glucose nanofibers
The SEM images in Fig. 1 show that the morphology and size of PVA/HA/glucose nanofibers are strongly dependent on glucose contents. At high glucose content (HA/glucose = 1:10), the nanofibers presented an unsmooth surface and an average diameter at 450 ± 10 nm [Fig. 1a]. With decreasing glucose contents, the nanofibers showed a continuous and uniform surface with a decrease in average diameter [300 ± 10 nm for HA/glucose = 1:5, Fig. 1b; 250 ± 8 nm for HA/glucose = 1:3, Fig. 1c]. Fig. S3 (ESI†) shows that the viscosities of electrospinning solutions increased with increasing glucose contents (i.e., decreasing HA/glucose ratios). The lower viscosity resulted in the easy splitting of droplets and the fibers became thinner. Especially, as HA/glucose ratio reached 1:1, the fibers became uneven and interspersed with beads and the diameter of fibers decreased to 140 ± 4 nm [Fig. 1d]. This is due to surface tension effects may dictate fiber formation ability at low concentrations with low solution viscosity and droplets or microbeads will form.41,42 Therefore, a representative HA/glucose ratio at 1:3 was used to fabricate the PVA/HA/glucose nanofibers to prepare meso-HA/AC nanofibers as precursors.
 |
| | Fig. 1 SEM images of PVA/HA/glucose nanofibers with HA/glucose ratios at (a) 1:10, (b) 1:5, (c) 1:3, and (d) 1:1. | |
3.2 Effects of hydrothermal temperature and time on morphology of nanofibers
Fig. 2 illustrates the SEM images of representative PVA/HA/glucose nanofibers (HA/glucose = 1:3, 250 ± 8 nm) at different hydrothermal temperatures for 16 h. Fig. 2a shows the diameter of the nanofibers significantly decreases to 180 ± 9 nm under 160 °C for 16 h as compared to precursor, however, the diameter of the nanofibers is still larger as compared to the nanofibers under 180 °C for the same time (Fig. 2b, 18 ± 2 nm), owing to the low carbonization of the polymeric matrix. In addition, as the temperature was raised to 200 °C, the sketch of fibers tended to be obscure (Fig. 2c), suggesting that fibrous structure had been destroyed in such a high temperature. Therefore, a temperature of 180 °C was determined to be the optimum hydrothermal temperature.
 |
| | Fig. 2 SEM images of PVA/HA/glucose nanofibers at different hydrothermal temperatures: (a) 160 °C, (b) 180 °C, and (c) 200 °C (HA/glucose = 1:3, 16 h). | |
For evaluation of hydrothermal time on the morphology of nanofibers, the PVA/HA/glucose nanofibers were hydrothermal treated under 180 °C for four times (12, 16, 24, and 48 h, Fig. 3). After hydrothermal treated at 180 °C for 12 h, the diameter of fibers significantly decreased to 50 ± 2 nm, meanwhile, the carbon beads were forming along fibers, as a result, an interesting surface with bead-on-string structure was presented (Fig. 3a). As prolonging time to 16 h, the fibers could still hold the bead-on-string structure and aggregated together to give the slit-shaped pores, nevertheless, the diameter of fiber further decreased to 18 ± 2 nm, and the carbon beads were about 20 ± 1 nm in diameter (Fig. 3b). It was speculated that the bead-on-string structure was much like “–carbon bead–HA rod–carbon bead–”, which was further confirmed by the following TEM and XRD analysis. As further prolonging time, the fiber shape was damaged completely. The fibers have changed into leaf-shaped (Fig. 3c, 24 h) and petal-shaped (Fig. 3d, 48 h). A probable reason is that the sufficient growth of HA crystals broke the bondage of fiber and showed leaf-shaped or petal-shaped morphology. Therefore, 16 h was selected for the preparation of meso-HA/AC nanofibers.
 |
| | Fig. 3 SEM images of PVA/HA/glucose nanofibers at different hydrothermal time: (a) 12 h, (b) 16 h, (c) 24 h, (d) 48 h (HA/glucose = 1:3, 180 °C); TEM images of meso-HA/AC nanofibers (e) and (f) magnification for (e) (HA/glucose = 1:3, 180 °C, 16 h). | |
Fig. 3e and f illustrates the TEM images of meso-HA/AC nanofibers (HA/glucose = 1:3, 180 °C, 16 h). As it could be seen from Fig. 3f, HA crystal showed a rod-like shape about 1 nm× 5 nm in diameter × length and assembled into bundle, which inserted into the two neighboring carbon beads to form the bead-on-string structure. XRD analysis was performed to confirm the crystallization of the meso-HA/AC nanofibers. Fig. 4A shows the typical diffraction peaks of hexagonal HA (space group P63/m) were presented at 2θ = 25.87° (002), 28.13° (102), 28.97° (210), 31.78° (211), 32.90° (300), 34.05° (202), 39.82° (310), 46.72° (222) and 49.48° (213), which could be indexed as the standard data (JCPDS no. 09-0432). In addition, a broad diffraction peaks about at 2θ = 20° revealed an amorphous carbon phase of carbon beads due to the carbonization of glucose.43 Fig. 4B shows the nitrogen adsorption and desorption isotherms for meso-HA/AC nanofibers. The isotherm of porous nanofibers was identified as typical IV-type curve with type H3 hysteresis loops (characteristic of mesoporous materials)44,45 attributed to the aggregation of bead-on-string structures giving rise to slit-shaped pores, which was in agreement with the results of SEM and TEM. Furthermore, the pores were mainly distributed around 29 nm (inset of Fig. 4B). The specific surface area of meso-HA/AC nanofibers was determined to be 30.014 m2 g−1, which was nearly four times larger than that of precursor PVA/HA/glucose nanofibers (5.875 m2 g−1).
 |
| | Fig. 4 (A) XRD pattern of meso-HA/AC nanofibers (HA/glucose 1:3, 180 °C, 16 h); (B) N2 adsorption–desorption isotherm for meso-HA/AC nanofibers, inset: pore size distribution of nanofibers based on nitrogen absorption; (C) FT-IR spectra of HA powders, glucose powders, and meso-HA/AC nanofibers; (D) Raman spectra of meso-HA/AC nanofibers. | |
3.3 FT-IR and Raman analysis
Fig. 4C shows FT-IR spectra of HA powders, glucose powders and meso-HA/AC nanofibers. In the spectrum of HA powders, the characteristic absorption peaks for HA were presented at: 3570 cm−1 (–OH stretching); 633 cm−1 (–OH deformation); 1041, 1091 cm−1 (v3 antisymmetric P–O stretching, threefold degenerated); 962 cm−1 (v1 symmetric P–O stretching) and 566, 604 cm−1 (v4 O–P–O bending, threefold degenerated). In addition, an obvious peak at 1380 cm−1 was attributed to residual nitrate groups resulting from synthesis precursors.46,47 In the spectrum of glucose powders, the characteristic absorption peaks for glucose were at: 3374 cm−1 (broad, –OH stretching); 1620 cm−1 (–OH bending); 2930 and 2855 cm−1 (–CH2, –CH stretching), 1436, 1378 and 1318 cm−1 (C–H bending); 1701 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), 1050 and 1008 cm−1 (C–O stretching). Compared with the spectra of HA and glucose, the characteristic absorption peaks for both HA and glucose were presented in the spectrum of meso-HA/AC nanofibers, however, the characteristic peak of –OH at about 3374 cm−1 (especially at 1620 cm−1) was significantly weakened due to the carbonization of glucose. Meanwhile, the strong band at 1585 cm−1 and a shoulder at 1385 cm−1 in Fig. 4D are attributed to the in-plane vibration associated with graphite carbon (G band), and a broad lower frequency band is assigned to disordered amorphous carbon (D band).48 On the basis of FT-IR and Raman analysis results, it can be speculated that carbon beads were activated carbon (AC) structured with CO and –OH groups.
O stretching), 1050 and 1008 cm−1 (C–O stretching). Compared with the spectra of HA and glucose, the characteristic absorption peaks for both HA and glucose were presented in the spectrum of meso-HA/AC nanofibers, however, the characteristic peak of –OH at about 3374 cm−1 (especially at 1620 cm−1) was significantly weakened due to the carbonization of glucose. Meanwhile, the strong band at 1585 cm−1 and a shoulder at 1385 cm−1 in Fig. 4D are attributed to the in-plane vibration associated with graphite carbon (G band), and a broad lower frequency band is assigned to disordered amorphous carbon (D band).48 On the basis of FT-IR and Raman analysis results, it can be speculated that carbon beads were activated carbon (AC) structured with CO and –OH groups.
3.4 Sorption experiments
3.4.1 Effect of pH and ionic strength. The hydrolysis constants of Co(II) (logK1 = −9.6, logK2 = −9.2, and logK3 = −12.7)49 demonstrate that Co(II) presents in the form of Co2+, Co(OH)+, Co(OH)2 and Co(OH)3− at different pH values (Fig. S4, ESI†). Fig. 5 shows the sorption Co(II) onto meso-HA/AC nanofibers as a function of pH in NaNO3 solutions, which can be divided into three regions. The sorption percentage of Co(II) on meso-HA/AC in each given NaNO3 solutions increases slowly as the pH at pH < 7. In the region, the predominant species of Co(II) was Co2+ and the removal of Co(II) was mainly accomplished by sorption reaction, meanwhile, the protonation reaction on the surfaces of meso-HA/AC nanofibers (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SOH + H+ ↔ SOH2+) made it difficult for Co(II) to be adsorbed on and resulted in the low sorption efficiency of Co(II) due to the electrostatic repulsion. However, the nanofibers still showed relatively higher sorption percentage (i.e. the percentage was increasing from 16.3% to 42% at pH 2.5–7) because AC beads structured many CO groups on surface besides –OH groups. On sharp contrast to region I, the sorption percentage increases significantly at pH 7–10. In this region, the predominant species of Co(II) were Co(OH)+ and the concentration of deprotonated sites (SO−) increased with increasing pH because of the surface deprotonation reaction (SOH ↔ SO− + H+), which enhanced the sorption of the positively charged Co(II) ions through electrostatic attraction.50 At pH > 10, the sorption percentage increases unobviously and maintains a high value above 95% [predominant species of Co(II) were Co(OH)2].
SOH + H+ ↔ SOH2+) made it difficult for Co(II) to be adsorbed on and resulted in the low sorption efficiency of Co(II) due to the electrostatic repulsion. However, the nanofibers still showed relatively higher sorption percentage (i.e. the percentage was increasing from 16.3% to 42% at pH 2.5–7) because AC beads structured many CO groups on surface besides –OH groups. On sharp contrast to region I, the sorption percentage increases significantly at pH 7–10. In this region, the predominant species of Co(II) were Co(OH)+ and the concentration of deprotonated sites (SO−) increased with increasing pH because of the surface deprotonation reaction (SOH ↔ SO− + H+), which enhanced the sorption of the positively charged Co(II) ions through electrostatic attraction.50 At pH > 10, the sorption percentage increases unobviously and maintains a high value above 95% [predominant species of Co(II) were Co(OH)2].
 |
| | Fig. 5 Effect of ionic strength and pH on sorption of Co(II) onto meso-HA/AC nanofibers. T = 303.15 K, CCo(initial) = 1.67 × 10−4 mol−1 L, m/V = 0.8 g L−1. | |
It can also be seen from Fig. 5 that the sorptions of Co(II) onto meso-HA/AC nanofibers are strongly dependent on NaNO3 concentrations at pH < 10. As the electrolyte concentration decreased, the electrical double layer complexes formed by Co(II) ions and nanofibers would favor the sorption. Meanwhile, the ionic strength of solution influenced the activity coefficient of metal ions, which limited the transfer to the composite surfaces. Generally, outer-sphere surface complexation and cation exchange are more vulnerable to ionic strength variations than inner-sphere surface complexation. Therefore, the removal was dominated by ion exchange and outer-sphere surface complexation at pH < 10; the no distinguish in Co(II) removal at pH > 10 indicates that innersphere surface complexation is the main sorption mechanism at high pH values.51
3.4.2 Sorption kinetics. Fig. 6A shows the sorptions of Co(II) on sorbents as a function of contact time. The sorption of Co(II) on meso-HA/AC nanofibers increased sharply in the first contact time of 400 min, after then the sorption rate was gradually slowed down. In the first 400 min, the physisorption of Co(II) on sorbent by intermolecular forces was predominant, which was faster than chemisorption (mainly in the form of ion exchange); after then, the physisorption was weakened, while the chemisorption was enhanced, correspondingly, the sorption rate was gradually slowed down. After 1600 min, the adsorption equilibrium was established.
 |
| | Fig. 6 (A) Effect of contact time on the sorption of Co(II) onto meso-HA/AC nanofibers and (B) W–M kinetic model of the sorption of Co(II) onto meso-HA/AC. T = 303.15 K, pH = 6.5 ± 0.1, I = 0.01 mol L−1 NaNO3, CCo(initial) = 1.67 × 10−4 mol L−1, m/V = 0.8 g L−1. | |
In order to evaluated the kinetic parameters of Co(II) sorption onto meso-HA/AC nanofibers, the pseudo-second-order kinetic model52 and Weber and Morris kinetic (W–M) model53 are employed to simulate the kinetic sorption:
| |
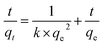 | (3) |
where
qt (mg g
−1) is the amount of Co(
II) adsorbed on the surface of meso-HA/AC nanofibers at time
t (min),
qe (mg g
−1) is the equilibrium sorption capacity,
k (g mg
−1 min
−1) is the rate constant of pseudo-second-order kinetics,
ki (mg g
−1 min
−1/2) is intra particle diffusion rate constant of Weber and Morris, and
c (mg g
−1) is a constant.
The simulate result of pseudo-second-order model is shown in Fig. 6A (inset), the high correlation coefficient (R2 = 0.99855) implies that the pseudo-second-order model is valid to describe the sorption of Co(II) onto meso-HA/AC nanofibers. Fig. 6B illustrates the simulate result of Weber and Morris model. As seen from Fig. 6B, the relation plot of qt and t1/2 does not go through the whole origin, instead, two better linear relationships were obtained for the two stages (first one: R2 = 0.99561, second one: R2 = 0.96487), moreover, the slope of the linear line for the first stage was steeper than that of the second one. The two interdependent stages indicate that intraparticle diffusion is not the sole rate-controlling step in the sorption process of Co(II) onto the meso-HA/AC nanofibers.54 The initial rapid uptake can be attributed to the boundary layer effects. After the external surface loading completes, the intraparticle diffusion takes place.25
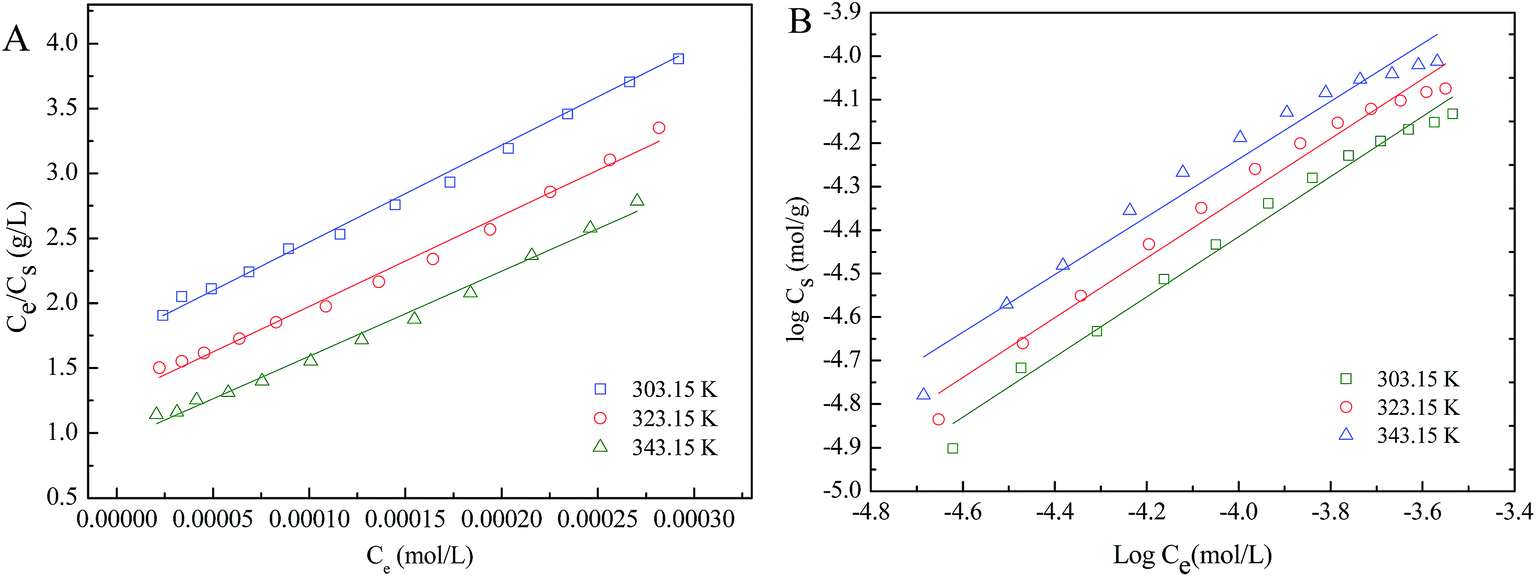
3.4.3 Sorption isotherms. The sorption isotherms of Co(II) at three different temperatures show that the sorption of Co(II) increases with increasing temperature (Fig. 7). In order to further describe the sorption of Co(II) onto nanofibers, two well known models of Freundlich and Langmuir isotherms are used to fit the equilibrium data. The Langmuir isotherm model is normally used to analyze monolayer sorption on a uniform surface,55 which can be expressed in linear form:| |
 | (5) |
where Cs max is the maximum sorption capacity at complete monolayer coverage (mol g−1), and b (L mol−1) is a constant associated with the sorption energy. Ce is the equilibrium concentration of metal ions remained in the solution (mol L−1). Cs is the amount of metal ions adsorbed on per weight unit of solid after equilibrium (mol g−1). On the other hand, the Freundlich model assumes a homogeneous surface56 and can be expressed in linear form:| | |
logCs = logkF + nlogCe
| (6) |
where kF (mol1−n Ln g−1) represents the sorption capacity when metal ion equilibrium concentration equals to 1, and n represents the degree of dependence of sorption with equilibrium concentration.
 |
| | Fig. 7 Sorption isotherms of Co(II) onto meso-HA/AC nanofibers at three different temperatures. pH = 6.5 ± 0.1, I = 0.01 mol L−1 NaNO3, m/V = 0.8 g L−1. | |
The relative parameters calculated on the basis of Langmuir and Freundlich models (Fig. 8) are listed in Table 1. The two models can well fit the sorption isotherms (R2 > 0.96), and Langmuir model is slightly better than Freundlich model. The sorption of Co(II) according with Langmuir isotherm demonstrates that the whole surface of nanofibers has identical sorption activity, which is beneficial to forming almost complete monolayer coverage. The values of Cs max calculated by Langmuir model for sorption of Co(II) onto sorbent increases with increasing temperature, suggesting that high temperature is beneficial to sorption process. Additionally, each value of n obtained from the Freundlich model is lower than 1, indicating that a nonlinear sorption has taken place on the surface of nanofibers. The meso-HA/AC bead-on-string nanofibers showed slightly better sorption capacity than that of the meso-HA nanofibers reported in previous work [Table S1 (ESI†)].
 |
| | Fig. 8 (A) Langmuir and (B) Freundlich isotherm for Co(II) sorption onto meso-HA/AC nanofibers at three different temperatures. pH = 6.5 ± 0.1, I = 0.01 mol L−1 NaNO3, m/V = 0.8 g L−1. | |
Table 1 Parameters for Langmuir and Freundlich isotherm models for the sorption of Co(II) onto meso-HA/AC nanofibers
| Models |
Parameters |
343.15 K |
323.15 K |
303.15 K |
| Langmuir |
Cs max (mol g−1) |
1.53 × 10−4 |
1.44 × 10−4 |
1.40 × 10−4 |
| b (L mol−1) |
7.00 × 103 |
5.49 × 103 |
4.11 × 103 |
| R2 |
0.9910 |
0.9903 |
0.9935 |
| Freundlich |
kF (mol1−n Ln g−1) |
2.61 × 10−2 |
2.51 × 10−2 |
2.23 × 10−2 |
| n |
0.663 |
0.686 |
0.691 |
| R2 |
0.9638 |
0.9772 |
0.9856 |
4. Conclusions
In the present work, a representative HA/glucose ratio at 1:3 was used to fabricate the PVA/HA/glucose nanofibers as precursors to prepare the resulting meso-HA/AC bead-on-string nanofibers by hydrothermal process. The nanofibers are confirmed to be mesoporous characteristic attributed to the aggregation of bead-on-string structures. Herein, the pores were mainly distributed around 29 nm, and the specific surface area was 30.014 m2 g−1. Additionally, HA crystal showed a rod-like shape about 1 nm × 5 nm in diameter × length and assembled into bundle, which inserted into the two neighboring carbon beads to form the bead-on-string structure, and the carbon beads were activated carbon structured with CO and –OH groups.
The sorption of Co(II) onto meso-HA/AC nanofibers in each given NaNO3 solution increases slowly as the pH at pH < 7, significantly at pH 7–10 before maintains a high value above 95%. The sorptions of Co(II) onto the meso-HA/AC nanofibers are strongly dependent on NaNO3 concentrations at pH < 10, no distinguish at pH > 10. The pseudo-second-order model is valid to describe the sorption of Co(II) onto meso-HA/AC nanofibers, and intra particle diffusion is not the sole rate-controlling step. Both Langmuir and Freundlich models can well describe the equilibrium data of Co(II) ions sorption, and Langmuir model is slightly better than Freundlich model. Moreover, the thermodynamic parameters imply that the sorption process is spontaneous and endothermic. The meso-HA/AC bead-on-string nanofibers may have potential as a high effective material for the removal of heavy metal ions from aqueous solution.
Acknowledgements
Financial support from National Natural Science Foundation of China (31371859 and 31171788) is gratefully acknowledged.
References
- H. Wang, P. Zhang, X. Ma, S. Jiang, Y. Huang, L. Zhai and S. Jiang, J. Hazard. Mater., 2014, 265, 158–165 CrossRef CAS PubMed.
- M. Aliabadi, M. Irani, J. Ismaeili, H. Piri and M. J. Parnian, Chem. Eng. J., 2013, 220, 237–243 CrossRef CAS.
- S. S. Gupta and K. G. Bhattacharyya, Phys. Chem. Chem. Phys., 2012, 14, 6698–6723 RSC.
- S. Zhang, Z. Guo, J. Xu, H. Niu, Z. Chen and J. Xu, J. Radioanal. Nucl. Chem., 2011, 288, 121–130 CrossRef CAS.
- R. W. Leggett, Sci. Total Environ., 2008, 389, 259–269 CrossRef CAS PubMed.
- D. Manohar, B. Noeline and T. Anirudhan, Appl. Clay Sci., 2006, 31, 194–206 CrossRef CAS.
- M. Abbas, S. Kaddour and M. Trari, J. Ind. Eng. Chem., 2014, 20, 745–751 CrossRef CAS.
- E. Oguz and M. Ersoy, Ecotoxicol. Environ. Saf., 2014, 99, 54–60 CrossRef CAS PubMed.
- G. Chaudhuri, P. Dey, D. Dalal, P. Venu-Babu and W. R. Thilagaraj, Water, Air, Soil Pollut., 2013, 224, 1–11 CrossRef CAS.
- M. M. Matlock, B. S. Howerton, J. D. Robertson and D. A. Atwood, Ind. Eng. Chem. Res., 2002, 41, 5278–5282 CrossRef CAS.
- A. Smara, R. Delimi, E. Chainet and J. Sandeaux, Sep. Purif. Technol., 2007, 57, 103–110 CrossRef CAS.
- M. Gamal Khedr, Desalin. Water Treat., 2009, 2, 342–350 CrossRef.
- K. Hassan, J. Radioanal. Nucl. Chem., 2011, 289, 801–804 CrossRef CAS.
- C. Blöcher, J. Dorda, V. Mavrov, H. Chmiel, N. Lazaridis and K. Matis, Water Res., 2003, 37, 4018–4026 CrossRef.
- B. R. Reddy and D. N. Priya, J. Power Sources, 2006, 161, 1428–1434 CrossRef CAS.
- C. Chen and X. Wang, Appl. Geochem., 2007, 22, 436–445 CrossRef CAS.
- S. Wang, J. Hu, J. Li and Y. Dong, J. Hazard. Mater., 2009, 167, 44–51 CrossRef CAS PubMed.
- L. Zhang, L. Huang, Z. Zeng, J. Qian and D. Hua, Phys. Chem. Chem. Phys., 2016, 18, 13026–13032 RSC.
- W. Song, X. Wang, Q. Wang, D. Shao and X. Wang, Phys. Chem. Chem. Phys., 2015, 17, 398–406 RSC.
- W. Zheng, X.-m. Li, F. Wang, Q. Yang, P. Deng and G.-m. Zeng, J. Hazard. Mater., 2008, 157, 490–495 CrossRef CAS PubMed.
- M. J. Angove, B. B. Johnson and J. D. Wells, J. Colloid Interface Sci., 1998, 204, 93–103 CrossRef CAS PubMed.
- Ö. Yavuz, Y. Altunkaynak and F. Güzel, Water Res., 2003, 37, 948–952 CrossRef PubMed.
- Ş. Kubilay, R. Gürkan, A. Savran and T. Şahan, Adsorption, 2007, 13, 41–51 CrossRef.
- M. L. Schlegel, L. Charlet and A. Manceau, J. Colloid Interface Sci., 1999, 220, 392–405 CrossRef CAS PubMed.
- I. Smičiklas, S. Dimović, I. Plećaš and M. Mitrić, Water Res., 2006, 40, 2267–2274 CrossRef PubMed.
- Y. Huang, L. Chen and H. Wang, J. Radioanal. Nucl. Chem., 2011, 291, 777–785 CrossRef.
- K. Swayampakula, V. M. Boddu, S. K. Nadavala and K. Abburi, J. Hazard. Mater., 2009, 170, 680–689 CrossRef CAS PubMed.
- V. Gopalakannan and N. Viswanathan, Ind. Eng. Chem. Res., 2015, 54, 12561–12569 CrossRef CAS.
- Q. Y. Ma, S. J. Traina, T. J. Logan and J. A. Ryan, Environ. Sci. Technol., 1994, 28, 1219–1228 CrossRef CAS PubMed.
- D. F. Mercado, G. Magnacca, M. Malandrino, A. Rubert, E. Montoneri, L. Celi, A. Bianco Prevot and M. C. Gonzalez, ACS Appl. Mater. Interfaces, 2014, 6, 3937–3946 CAS.
- S. J. Wu, F. T. Li, H. T. Wang, L. Fu, B. R. Zhang and G. T. Li, Polymer, 2010, 51, 6203–6211 CrossRef CAS.
- H. Wang, P. Zhang, X. Ma, S. Jiang, Y. Huang and L. Zhai, J. Hazard. Mater., 2014, 265, 158–165 CrossRef CAS PubMed.
- M. Irani, A. R. Keshtkar and M. A. Moosavian, Chem. Eng. J., 2012, 200, 192–201 CrossRef.
- S. Madhugiri, B. Sun, P. G. Smirniotis, J. P. Ferraris and K. J. Balkus, Microporous Mesoporous Mater., 2004, 69, 77–83 CrossRef CAS.
- R. Cai, H. Wang, M. Cao, L. Hao, L. Zhai, S. Jiang and X. Li, Mater. Des., 2015, 87, 17–24 CrossRef CAS.
- X. Dong, J. Wang, Q. Cui, G. Liu and W. Yu, Int. J. Chem., 2009, 1, 13–17 CAS.
- X. Dong, J. Wang, Q. Cui, G. Liu and W. Yu, Mod. Appl. Sci., 2009, 3, 75–80 CAS.
- S. Asuha, X. Zhou and S. Zhao, J. Hazard. Mater., 2010, 181, 204–210 CrossRef CAS PubMed.
- L. Zhang, W. Zhu, H. Zhang, S. Bi and Q. Zhang, RSC Adv., 2014, 4, 30542–30550 RSC.
- L.-C. Lin, M. Thirumavalavan, Y.-T. Wang and J.-F. Lee, Colloids Surf., A, 2010, 369, 223–231 CrossRef CAS.
- J. Deitzel, J. Kleinmeyer, D. Harris and N. B. Tan, Polymer, 2001, 42, 261–272 CrossRef CAS.
- Y. P. Neo, S. Ray, A. J. Easteal, M. G. Nikolaidis and S. Y. Quek, J. Food Eng., 2012, 109, 645–651 CrossRef CAS.
- R. Cakan, Chem. Commun., 2008, 3759–3761 RSC.
- S. Yang, J. Li, D. Shao, J. Hu and X. Wang, J. Hazard. Mater., 2009, 166, 109–116 CrossRef CAS PubMed.
- Q. Dong, H. Su, D. Zhang, Z. Liu and Y. Lai, Microporous Mesoporous Mater., 2007, 98, 344–351 CrossRef CAS.
- S. Raynaud, E. Champion, D. Bernache-Assollant and P. Thomas, Biomaterials, 2002, 23, 1065–1072 CrossRef CAS PubMed.
- A. Rajendran, R. C. Barik, D. Natarajan, M. Kiran and D. K. Pattanayak, Ceram. Int., 2014, 40, 10831–10838 CrossRef CAS.
- C. Yao, Y. Shin, L.-Q. Wang, C. F. Windisch, W. D. Samuels, B. W. Arey, C. Wang, W. M. Risen and G. J. Exarhos, J. Phys. Chem. C, 2007, 111, 15141–15145 CAS.
- H. Yüzer, M. Kara, E. Sabah and M. S. Çelik, J. Hazard. Mater., 2008, 151, 33–37 CrossRef PubMed.
- J. Hu, C. Chen, X. Zhu and X. Wang, J. Hazard. Mater., 2009, 162, 1542–1550 CrossRef CAS PubMed.
- S. Yang, G. Sheng, X. Tan, J. Hu, J. Du, G. Montavon and X. Wang, Geochim. Cosmochim. Acta, 2011, 75, 6520–6534 CrossRef CAS.
- Y.-S. Ho and G. McKay, Chem. Eng. J., 1998, 70, 115–124 CrossRef CAS.
- W. J. Weber and J. C. Morris, J. Sanit. Eng. Div., 1963, 89, 31–60 Search PubMed.
- S. R. Popuri, Y. Vijaya, V. M. Boddu and K. Abburi, Bioresour. Technol., 2009, 100, 194–199 CrossRef CAS PubMed.
- Y. Takahashi, Y. Minai, S. Ambe, Y. Makide and F. Ambe, Geochim. Cosmochim. Acta, 1999, 63, 815–836 CrossRef CAS.
- M. Kilpatrick, L. L. Baker Jr and C. D. McKinney Jr, J. Phys. Chem., 1953, 57, 385–390 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra14193e |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.