
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanostructure and pore size control of template-free synthesised mesoporous magnesium carbonate†
Ocean
Cheung
a,
Peng
Zhang
a,
Sara
Frykstrand
a,
Haoquan
Zheng
b,
Taimin
Yang
b,
Marco
Sommariva
c,
Xiaodong
Zou
b and
Maria
Strømme
*a
aDivision for Nanotechnology and Functional Materials, Department of Engineering Sciences, Uppsala University, Uppsala, SE-751 21, Sweden. E-mail: maria.stromme@angstrom.uu.se
bDepartment of Materials and Environmental Chemistry, Stockholm University, Stockholm, SE-10691, Sweden
cPANalytical B.V., Lelyweg 1 (7602 EA), PO Box 13, Almelo, 7600 AA, The Netherlands
First published on 1st August 2016
Abstract
The structure of mesoporous magnesium carbonate (MMC) first presented in 2013 is investigated using a bottom-up approach. MMC is found to be built from the aggregation of nanoparticles of amorphous MgCO3 and MgO with a coating of amorphous MgCO3. The nanoparticles have dimensions of approximately 2–5 nm as observed using transmission electron microscopy and the aggregation of the particles creates the pore structure of MMC. We further show that the average pore diameter of MMC can be controlled by varying the temperature during the powder formation process and demonstrate that altering the pore size opens the possibility to tune the amorphous phase stabilisation properties that MMC exerts on poorly soluble drug compounds. Specifically, we show the loading and release of the antifungal drug itraconazole using MMC as a drug carrier.
Introduction
After the invention of the scanning tunnelling microscope in 1981, the discovery of the fullerenes in 1985, and Drexler's dystopian presentation of nanotechnology in his 1986 book Engines of Creation,1 nanomaterials have made their entrance in most of the materials development industries.An important class of nanomaterials is nanoporous materials. Nanoporous materials are constituted by micro- and mesoporous materials. The most well-known microporous materials (average pore size < 2 nm) are probably the zeolites. Significant efforts have been spent on developing these crystalline framework aluminosilicate materials for gas separation and catalysis. In particular, the pore apertures on some zeolites were found to be adjustable. The pore apertures (or the pore windows) can be controlled by performing different types of post-synthesis treatments, such as cation exchange,2–4 heat/vacuum treatment or dehydration.5,6
Whilst the narrow pores of zeolites make them very desirable in applications related to small molecules, a number of different types of mesoporous materials (average pore size ∼ 2–50 nm) are presently being developed for applications involving larger molecules. These mesoporous materials include silica, alumina, titania and carbon.7–12 Of these, mesoporous silica materials are the most studied. The formation of mesoporous silica is well understood,13–19 and the application of mesoporous silica has reached the industrial implementation stage (e.g. in chromatography).20 Good understanding of the reaction mechanism and the final structure are essential for the development of mesoporous materials towards industrial applications. The synthesis of mesoporous silica involves the replication of a surfactant liquid crystal structure and the polymerisation of a silica precursor. Removal of the organic surfactant leads to a porous structure that is supported by a hard silica framework.
Due to its large variation abilities, mesoporous silica has been evaluated in a number of applications ranging from drug delivery,21–27 regeneration of bone tissue28,29 and as vaccine adjuvants,30,31 to catalysis and as adsorbents.32–36 This material can also be functionalised by amine grafting, making it potentially suitable as adsorbent for applications such as CO2 separation.37
An important aspect regarding the practical usefulness of mesoporous materials is the ability to precisely tailor the pore size for a specific application. Different methods to control the pore size of mesoporous silica have been developed.38 These methods include using specially selected organic template/surfactants39,40 and the addition of a swelling agent41,42 in the synthesis. Another pore size control method is post synthesis functionalisation of the material using amines.43 Up to now, accurate pore size control of mesoporous silica can only be achieved with the use of additional organic reagents during or post synthesis.
Currently there are many different types of magnesium compounds that are being tested for applications such as CO2 capture and storage,44,45 the removal of hazardous materials46 and drug delivery.22,23 Previously, we presented the template-free synthesis of a mesoporous magnesium carbonate (MMC) material with high surface area (up to 800 m2 g−1).47 The pore size distribution of the materials (which has been commercialised under the name Upsalite®) was relatively narrow and was size around 5 nm in diameter.47,48 This makes MMC a comparable material to mesoporous silica.
One may expect that such surfactant-free synthesis routes, where calcination at high temperatures is not needed, may be important for the up-scalable production of mesoporous materials. The industrial feasibility of the MMC is further emphasised by the material's moisture sorption properties,47–50 biocompatibility51 and drug stabilising properties.22,23 Most recently we also showed that MMC has the ability to prevent blood clotting,52 which opens up its potential for application where blood clotting needs to be suppressed. In order to further develop MMC and other mesoporous materials that are synthesised in surfactant-free processes towards novel applications areas, it is crucial to have a detailed understanding of the structure. Such understanding will allow further engineering and control of the material, such as pore size optimisation and surface functionalisation.
Our previous study showed that the pores were created in a two-step process including the formation of nanopores during solvent evaporation and subsequently the release of physically bound carbon dioxide (CO2).47,48 In this study, we present our latest results from our investigation on MMC. Specifically we reveal the detailed nanostructure of MMC using a bottom-up approach starting at nm scale. This has not been possible previously due to the large quantity of unreacted MgO in the resulting material. The material we presented in 2013 had an average pore size of around 5 nm. Here, we describe in detail the particle formation step of MMC and how the energy input during certain stages of the synthesis can be manipulated to accurately control the average pore size of MMC from 3 to 20 nm. We show that the difference in pore size on MMC can be used to control the enhanced release rate of a poorly soluble drug – itraconazole.
Experimental
Optimised synthesis of transparent mesoporous magnesium carbonate (MMC)
The typical synthesis procedure of transparent MMC includes the three steps (i) sol–gel synthesis, (ii) powder formation and (iii) degassing as described below. Note that the chemical composition of the reaction gel, and the synthesis/drying steps, are adapted and optimised from our previous studies. | ||
| Fig. 1 (a) The appearance of the material reaction mixture at different stages (1) reaction mixture after centrifugation, (2) and (3) reaction mixture forming a gel, (4) wet powder dried at room temperature and (5) dried transparent powder dried at 80 °C and then 150 °C and finally at 250 °C (b and c) transparent powder. | ||
Powder X-ray diffraction (PXRD)
PXRD analysis was conducted by a Bruker D8 Advanced XRD (Bruker, Bremen, Germany) Twin-Twin instrument (45 kV and 40 mA) with CuKα radiation (λ = 0.154 nm) in the 2θ range from 10° to 80°, with a step size of 0.02° and a measuring time of 2 s per step. Silicon zero background sample holders were used.Dynamic light scattering (DLS)
DLS was performed on a Malvern Instruments ZetaSizer NanoZS instrument (Malvern, UK). The diluted centrifuged liquid reaction mixtures were rapidly pipetted into a cuvette (1 cm path length) and added to the DLS apparatus. All statistics reported are the mean and standard deviation of measurements on triplicate samples.Thermogravimetric analysis (TGA)
TGA analysis was carried out using a Mettler Toledo TGA 2 instrument (Greifensee, Switzerland). The samples were heated in air from room temperature (RT) to 850 °C with a heating rate of 5 °C min−1.Scanning electron microscopy (SEM)
SEM images were recorded using a Zeiss LEO 1550 scanning electron microscope (Oberkochen, Germany) operating at 1 kV. Specimens were mounted on aluminium stubs with double sided carbon tape and coated with Pd/Au prior to analysis to avoid charging effects. Additional SEM images (Fig. S2†) were recorded using a Hitachi TM3000 table top SEM instrument (Tokyo, Japan) operating at 15 kV.X-ray photoelectron spectroscopy (XPS)
XPS was conducted on a PHI Quantum 2000 Scanning ESCA microprobe (Chanhassen, USA). Prior to analysis, the samples were sputter cleaned using argon ions for 10 min at 200 V in order to remove surface adsorbed contaminations. A full spectrum was recorded together with energy resolved spectra for Mg2p and O1s. During data acquisition, an electron beam of 20 mA was used together with argon ions to neutralise the non-conducting sample. The peak fittings were made with CasaXPS software, the curves were fitted using Gaussian–Lorentzian functions and the background was subtracted using a Shirley function. The spectra were calibrated against the O1s peak for magnesium oxide (531.0 eV) instead of the C1s peak at 285.0 eV for adventitious carbon that is commonly used as a reference. However, in the case of MgO, the binding energy for adventitious carbon is not reliable as reference. This is because the hydrocarbons interact with MgO in a way that shifts the C1s peak randomly, making the C1s peak unsuitable as a reference. Therefore, the O1s peak for MgO (531.0 eV) was used as an internal reference.53Total X-ray scattering and pair distribution function (PDF) analysis
Total scattering measurements were performed on an PANalytical Empyrean multipurpose diffractometer (Almelo, the Netherlands) equipped with a sealed high resolution X-ray tube with a silver anode (Ag K-alpha characteristic radiation λ = 0.5609 Å). The sample was loaded in a glass capillary with 2.0 mm external diameter. An empty capillary of the same type was measured in the same way for background subtraction. The measurements were performed using a setup with a focusing X-ray multilayer mirror on the incident beam side, and a solid state GaliPIX3D detector on the diffracted beam side. Anti-scatter kit specifically developed for the PDF application was used to achieve the needed featureless background.54–56 The data was recorded for 2θ between 3° and 148°, which corresponded to a Qmax value of 21.6 Å−1. An optimised variable counting time strategy was adopted to counteract the decrease of the scattered intensity at the highest angles due to the X-ray form factor.54–56 The total time of the measurement for both the sample and the empty capillary was 4 hours. The PANalytical HighScore Plus software57 was used for the background subtraction, the corrections and the normalisation needed for the generation of the experimental PDF from the measured data. PDFgui (V 1.0) was used for the analysis and fitting of the recorded data.58High resolution transmission electron microscopy (HRTEM)
HRTEM images were taken on a JEOL JEM-2100 FEG microscope (Tokyo, Japan) operating at 200 kV. Electron tomography tilt series were collected on JEOL JEM-2100 electron microscope. The tile angle was ranging from −63° to +66° with an interval of 1°. Before tomogram data collection, the samples were prepared by dispersing in ethanol and a droplet of the dispersion was placed on a copper grid (a droplet of 5 nm gold nanoparticle dispersion was also delivered on the copper grid to act as markers for tomogram alignment). The software RAPTOR59 was used for tilt series alignment. IMOD60,61 software was used for tomography reconstruction using the SIRT algorithm.IR spectroscopy
IR spectra were obtained using a Varian 610-IR FTIR (Santa Clara, California, United States) equipped with a Specac Goldengate attenuated total reflection accessory (Orpington, UK) with a diamond ATR element and KRS-5 lenses. The spectra (400–4000 cm−1) were recorded using a RT deuterated triglycine sulphate (DTGS) detector with 4 cm−1 resolution. 64 background spectra were recorded and accounted for. The samples spectra were signal averages of 128 scans.Nitrogen adsorption
The specific surfaces area and porosity of the transparent MMC were determined by recording N2 sorption isotherms using a Micromeritics ASAP 2020 surface area analysers (Norcross, GA, USA). Prior to analyses, the samples were pre-treated by heating to 373 K under dynamic vacuum (1 × 10−4 Pa) using a Micromeritic SmartVacPrep sample preparation unit. The equilibrium adsorption data point was obtained when the change in the pressure drops below 0.01% within a 10 s interval (with minimum 100 s delay).Elemental analysis
CHN analysis and ICP-OES experiments were performed by MEDAC Ltd, UK. For the ICP-OES experiments, solid samples were digested using nitric acid (HNO3).Drug loading and release
Three different concentrations of itraconazole (ITZ) (3Pharmaceutica, China) was loaded on to the synthesised powder by solvent evaporation and the actual amount loaded was confirmed by TGA while DSC was employed to analyse the crystalline state of the loaded drug. The samples were grounded to a fine powder and analysed by SEM (using Hitachi TM3000) before the drug release experiment (Fig. S2†) to ensure homogeneous particle size. The release of ITZ was measured using the USP-II dissolution method in Sotax AT7 Smart dissolution bath (Sotax AG, Aesch, Switzerland). Simulated gastric fluid was used as the drug release medium. Samples with a total drug loading of 0.25 mg ITZ were placed in vessels containing 1 L simulated gastric fluid (pH 1.3) under 37 °C and 50 rpm stir speed. UV/vis (UV-1800 Spectrophotometer, SHIMADZU, Japan) absorption at a wavelength of 263.1 nm was used to calculate the amount of ITZ released. Powder samples with three different average pore sizes (samples A, D and E in Table 1 as discussed later) were loaded with three different amounts of ITZ upon impregnation by a solution of methylene chloride containing 10 mg mL−1 of ITZ. To obtain a drug loading degree of 30, 45 and 60% (wt/wt), 0.5 g of transparent MMC was mixed with 21.5, 41 and 75 mL, respectively, of the drug containing solution. The mixture was left shaking for 24 hours at room temperature for loading to take place. After loading, the ITZ loaded powder samples were first heated to 45 °C for the solvent to evaporate, and thereafter dried at 85 °C for 24 hours. Before the drug release studies the samples were grounded into fine powder. The particles were found to be smaller than 100 μm as confirmed by the SEM images (Fig. S2†).| Sample | Powder formation temperature (°C) | BET surface area (m2 g−1) | Peak DFT pore size (nm) | Total pore volume (cm3 g−1) |
|---|---|---|---|---|
| A | −20 | 263 | ∼13 | 1.27 |
| B | 10 | 297 | ∼10 | 1.15 |
| C | 20 | 499 | ∼6.5 | 0.97 |
| D | 20 (no stirring) | 173 | ∼20 | 1.00 |
| E | 30 | 618 | ∼5.1 | 0.76 |
| F | 50 | 661 | ∼3.4 | 0.60 |
| G | 80 | 690 | ∼2.9 | 0.50 |
The actual loading percentages of the different powders were confirmed by TGA whereas DSC was performed to obtain information about the crystallinity of the drug inside the carrier powder. The DSC analysis was performed on a DSC Q2000 instrument from TA Instruments (New Castle, Delware, USA). Samples of 4.5–5.5 mg were weighed into 5 mm aluminium pans and sealed by a press. The samples were first cooled to −35 °C for stabilisation and then heated to 250 °C at a heating rate of 3 °C min−1.
Results and discussions
We have performed comprehensive materials characterisation using various techniques. We can now reveal the details of the structure of the MMC material starting at a nanometre scale. The additional MgO removal step we adopted in this study was an important part for the structural investigation. We will also discuss how the pore size of this material can be adjusted between ∼3 and ∼20 nm without the addition of organic templates/surfactants or swelling agents. MMC was found to be constructed from aggregated nanometre sized MgO nanocrystals (coated with amorphous with MgCO3) and amorphous MgCO3 nanoparticles. These aggregated nanocrystals and nanoparticles are adhered together by amorphous MgCO3 throughout.Structural characterisation of the as-synthesized MMC
MMC synthesised using the described procedures (with a 250 °C drying step) was X-ray amorphous and contained about 14–18 wt% MgO (and ∼82–86 wt% MgCO3) as determined by CHN elemental analysis, inductively coupled plasma optical emission spectrometry (ICP-OES) and TGA (see ESI Fig. S3 and Table S2†). This gave a stoichiometric MgO content of the transparent MMC of around 32–46 mol%. This portion of MgO could not be removed by centrifugation at 4969g. In our previous study, the MgO content on MMC was significantly higher than the MMC in this study (previously MgO content was around 50 wt%, compare to 14–18 wt% in this study).HRTEM images showed that on a nanometre scale, the transparent MMC powder particles consisted of aggregates of randomly oriented crystalline nanoparticles of ∼2–5 nm in size (Fig. 2b and c). For the discussion here, we refer to these 2–5 nm nanoparticles as Type I nanoparticles. The corresponding Fourier transform (FT) of the crystalline particles gives a ring-like pattern (Fig. 2d). The particles are identified from the d-spacings of the rings in the FT to be MgO. These d-spacings were similar to those obtained from electron diffraction patterns of the same sample that had been heat-treated to 800 °C to form well-defined large MgO crystals of 10–50 nm in size (ESI, inset of Fig. S4a†), as the powder material is known to decompose into MgO at high temperature. These MgO nanocrystals in MMC sometimes appeared aggregated and encapsulated by amorphous MgCO3, forming particles that may be up to 30 nm in size. We refer to these ∼30 nm particles as Type II nanoparticles in this discussion. The relationship between Type I and Type II nanoparticle is demonstrated by Fig. 3. Fig. 2e shows that a particle of MMC is made from aggregates of Type II nanoparticles. These Type II particles can be observed from the electron tomography using a tilt series of TEM images (Video V1, ESI†). Video V1† further confirms that the transparent MMC particles are constructed with aggregated particles of up to 30 nm in size (Type II).
 | ||
| Fig. 2 (a) 3D-Orthoslice representation of the reconstructed tomogram of MMC. (b) HRTEM image of a MMC particle showing randomly oriented MgO nanocrystals surrounded by amorphous. The crystalline parts are highlighted in red, (c) an enlarged view of a MgO nanocrystal marked in panel (b), (d) Fourier transform (FT) of the HRTEM image in panel (b) with the rings (marked) corresponding to MgO, (e) TEM image of a particle of MMC showing the aggregated nanoparticles. | ||
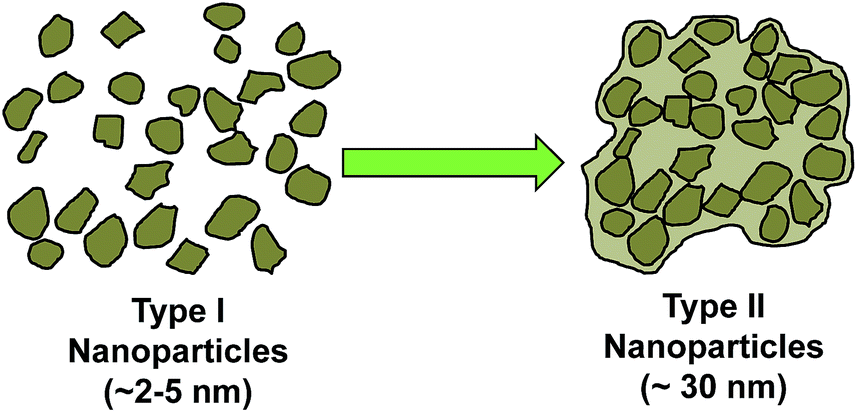 | ||
| Fig. 3 Schematic sketch of the formation of Type II nanoparticles from the aggregation of Type I MgO nanoparticles. | ||
When examined carefully, we identified two versions of Type II nanoparticles: the first (Type IIA) consists of aggregated Type I MgO nanoparticles of that were coated with a thin amorphous coating of MgCO3 on the surface. This was apparent due to an observable distinctive shape (which indicates MgO crystals) but with lack of sharp edges (which indicates that the surface of MgO crystals were covered with an amorphous layer) of some Type II particles, see Video V2 and Fig. S5† for more information. These amorphous regions were deduced to be MgCO3 by XPS and IR spectroscopy. Both techniques could only detect the presence of MgCO3 on the surface of the MMC particles (discussed later), suggesting that MgO was not exposed on the surface of MMC.
The second version of Type II nanoparticles (Type IIB) had no distinctive shapes and had smooth edges. These nanoparticles were concluded to be amorphous MgCO3 (by deduction from ICP-OES and TGA data) that were also up to ∼30 nm in size (those shown in Fig. 2e) and most probably formed from aggregation of Type I amorphous MgCO3 nanoparticles.
The presences of Type II particles, and how they aggregated together to form MMC particles, could also be seen using SEM (Fig. 4). Fig. 4a shows a typical MMC particle that macroscopically appeared to be smooth on the surface, but was in fact made from aggregated Type II nanoparticles that were up to 30 nm in size (Fig. 4b–d). The packing of these Type II particles appeared to be somewhat random but it was important to note that the images were in agreement with the tilt series TEM images shown in Video V1.† Note that the nanoparticles appear to have smooth and round surfaces, but this is mostly due to the thin coating applied on the sample, we therefore, could not draw any conclusions regarding the absolute morphologies of these nanoparticles (we also observed the presences of these nanoparticles on a sample without coating, however, these images are not shown here due to the significant charge build-up taking place when examining a sample of MMC without coating).
 | ||
| Fig. 4 (a–d) Scanning electron microscopy images of a particle of transparent MMC under different magnification, note the presence of nanoparticles (up to ∼30 nm in size) that make up a smooth particle of transparent MMC. | ||
The pore system of and the surface of MMC
The tomogram reconstructed from the title series (Video V2†) shows that the mesopores were present throughout the entire MMC particle as a result of nanoparticle/nanocrystal (Type I and Type II) aggregation (Fig. 2a, S5 and Video V2, ESI†). Both irregularly and regularly-shaped pores were observed, some of them were interconnected (Fig. S5†). The irregularly shaped pores were likely to have formed from the aggregation of Type I (∼2–5 nm) amorphous MgCO3 nanoparticles or Type I MgO nanocrystals coated with amorphous MgCO3. The regularly shaped pores may be a result of the aggregation of Type I MgO nanocrystals without or with very thin amorphous coating. In short, we could conclude that the MMC material originally presented in 2013 (ref. 47) was constructed from the hieratically aggregated nanoparticles; first, Type I nanoparticles with dimensions around 2–5 nm aggregate to form Type II nanoparticles of ∼30 nm in size, then the aggregation of Type II nanoparticles form the basis of the MMC material.We examined the transparent MMC using PXRD and found that the MgO nanocrystals (Type I nanoparticles) could not be detected by this analysis method due to their small crystal size, making the powder X-ray amorphous. The IR spectrum of the transparent MMC (ESI, Fig. S6 and S7†) showed that the IR bands related to MgCO3 were present. The Mg2p and O1s XPS spectra of the powder (ESI, Fig. S11†) showed that the surface of the powder particles contained mainly MgCO3 and a small amount of MgO. This further supported the idea that amorphous MgCO3 covered the aggregated nanocrystals/nanoparticles. Apart from the difference in the MgO content, the MMC in this study had no differences in the composition when compared with the materials presented in our previous studies (<20 wt%, compared to ∼50 wt% previously).22,23,47,50,51,62
Aggregate formation mechanism
In our previous studies, the formation mechanisms of the MMC47 and its pores48 were proposed. Further investigative work as that presented in this study provides a deeper understanding of the material's formation mechanism and ways to control such. The results from the various characterisation methods suggest that the material is formed by aggregation of different types of nanoparticles (Type I and Type II) in the reaction mixture containing also methanol with dissolved Mg2+ and CO32−. When methanol was evaporated from the reaction mixture, these aggregated nanoparticles were fixed in place, possibly by an additional layer of amorphous MgCO3 that precipitated from the reaction mixture upon drying (as illustrated by Fig. 5).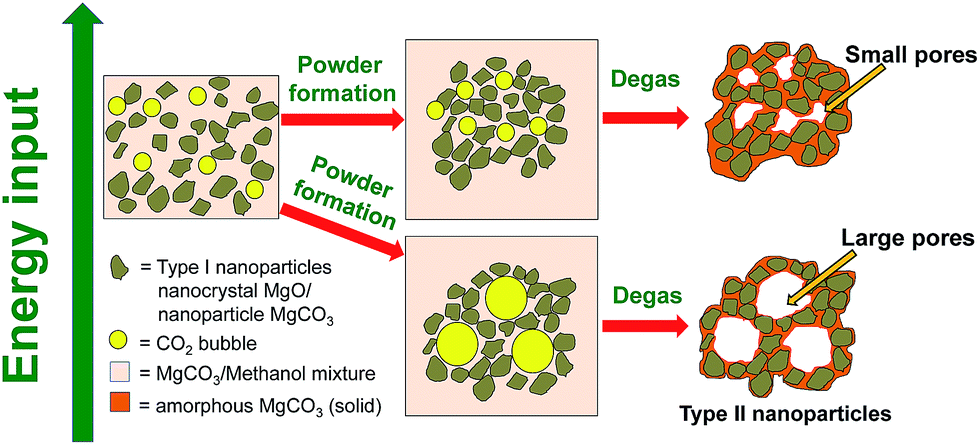 | ||
| Fig. 5 Schematic representation of the mechanism and energy input (e.g. temperature or stirring) dependence related to the pore formation of the mesoporous material. | ||
It was shown previously that MgO was practically insoluble in methanol (solubility ∼ 0.1%).63 The low solubility of MgO in methanol was also observed in this work, as indicated by the intense white cloudy mixture when the two were mixed. Interestingly, when the reaction vessel was pressurised with CO2 (4 bar) for 24 hours, the mixture became noticeably less cloudy, suggesting that some of the MgO particles had reacted or broken down into a more soluble compound. The reaction mixture became light yellow and only slightly cloudy, as observed earlier.47 After centrifugation, the reaction mixture is an optically clear, yellow solution. Tyndall scattering was observed by shining red laser light (650 nm) through the solution, indicating the presence of nanoparticles (ESI, Fig. S13†).
DLS was used to follow the behaviour of the observed nanoparticles upon gel drying. Nanoparticles of ∼50–100 nm in diameter were detected in the optically clear, yellow reaction mixture (ESI, Fig. S12†). Significant growth of these particles occurred with time when the reaction mixture was covered and left standing at room temperature (i.e. without active evaporation/drying). After 2 hours, the nanoparticles became too large to be detected by DLS. The observed particle growth was highly unlikely to be the growth of a single particle (as shown by HRTEM images and tomography study), but most probably the continued aggregation of Type II nanoparticles as described earlier.
The solid transparent form of the material was generated by particle aggregation during the powder formation step. A gel like material (Fig. 1a2 and a3) always appeared upon solvent evaporation before this gel collapsed into wet powder-like materials (Fig. 1a4). We followed the formation of the powder using IR spectroscopy (ESI, Fig. S8†) and the effect of additional water during this process (Fig. S9†). According to the IR spectra, no chemical reactions took place during the powder formation steps (from the liquid reaction mixture), only solvent evaporation was observed. IR bands related to MgCO3 were present in all of the recorded IR spectra. It is important to note that methyl magnesium carbonate has a similar yellow colour (in methanol or DMF) as our synthesis liquid and gel (Fig. 1a1–a3). Methyl magnesium carbonate is also known to decompose into amorphous MgCO3 (white), CO2 and methanol upon solvent evaporation via a gel like intermediate phase.64–66 IR spectroscopy could not distinguish methyl magnesium carbonate from an MgCO3/methanol mixture. Hence, as put forward earlier,47,48 it cannot be ruled out that that the nanoparticles detected by DLS is also amorphous MgCO3 nanoparticles that is produced from the decomposition of methyl magnesium carbonate upon gel drying.
Pore size control of MMC
The pore size distributions of materials that were synthesised using the different powder forming conditions outlined in Table 1 were obtained by density functional theory (DFT) analysis of N2 sorption isotherms (ESI, Fig. S14–20†). These transparent samples were prepared using various powder formation conditions (Table 1). The table also summarises the pore properties of the different materials showing that transparent MgCO3 powder could be made with very different pore sizes in a controlled manner.The average size of the mesopores could be controlled from ∼3 nm to ∼20 nm by adjusting the gel/powder formation rate in the powder formation step of the synthesis (Fig. 1). Using the information gathered in this study, we present Fig. 5 which shows a schematic sketch of how the pores in the material are formed. Fig. 5 also shows how the pore size can be controlled by adjusting the energy input during the powder formation step of the synthesis. Energy input, as written in Fig. 5, can be the use of heating, stirring or potentially any other changes to the system where energy is put into the system. In this study we focused on temperature being the variable means of energy whilst keeping the stirring rate constant at 75 rpm. This was because we found that temperature as a variable had given the most reproducible results when compared with other variables such as stirring speed.
During the powder formation step, a large amount of CO2 is given off from the reaction mixture. Both CO2 dissolved in methanol (in the reaction mixture) and possibly from the decomposition of methyl magnesium carbonate67 was eliminated. The eliminated gas phase CO2 molecules need to travel to the liquid/air interface between the reaction mixture/gel and ambient air before evaporating from the reaction mixture into gas phase. Stirring of the reaction mixture at a constant speed not only increases the material exchange at the interface between liquid and CO2 bubble, but also allows it to happen at a controlled (relatively constant) rate. However, when inside the reaction mixture, the CO2 molecules aggregate to form bubbles to reduce the surface energy. During the powder formation step, nanometre-sized particles (Type I and Type II nanoparticles) assembled around these CO2 bubbles which were then essentially trapped in this configuration. The nanometre sized particles also act as stabilisers for the CO2 bubbles.68 Low temperature allowed CO2 molecules to form aggregates in the reaction mixture at a higher extent than at high temperature (due to slower kinetics). The slow kinetics resulted in large bubbles and subsequently large pores. Sample D (Table 1) demonstrated the effect of reduced energy input (by eliminating stirring) clearly, as it had the largest pore size of the synthesised samples in this study. On the other hand, we found that varying the stirring rate (75 rpm and 125 rpm) within the limit of our setup can also vary the pore size slightly, but those results had poor reproducibility (discussed in ESI Fig. S21†) and the effect of stirring speed did not appear to be a dominating factor in pore size control in our setup.
After the wet powder forms (Fig. 1a4), the pores were fixed by heating under N2 flow at 85 °C. This step fixes the shape of the assembled powder particles and removes the trapped CO2 bubbles, resulting in a porous solid (Fig. 1a5).
Drug stabilisation using MMC
The impact of being able to control pore size of the synthesised material could be shown by its ability to stabilise amorphous compounds and the tailored drug release profile. The drug itraconazole (ITZ) was loaded into transparent MMC samples (the drug carrier) with three different average pore sizes (sample E, A and D with average pore sizes of ∼5.1 nm, ∼13 nm and ∼20 nm, respectively). ITZ, in its crystalline form, is a poorly water-soluble antifungal agent having a broad spectrum of activity against a variety of pathogens, including those most frequently causing opportunistic infection in human immunodeficiency virus (HIV) infected patients.69DSC was employed to analyse the structure of ITZ loaded into the pores of the carrier (ESI, Fig. S22†). TGA was used to confirm the concentration of drug loaded. The described drug loading procedures resulted in the different carrier samples being loaded with 30, 45 and 60 wt% of ITZ. An endothermic peak at 169 °C was observed in the DSC curve for the pure drug. This peak corresponds to the melting point of crystalline ITZ. For all carrier samples loaded with an ITZ concentration corresponding to 30%, this endothermic peak was absent. The lack of such peak confirmed that all carrier samples were able to completely suppress crystallisation of the drug loaded inside the pores. This observation was in line with earlier studies where ibuprofen was loaded in MMC22,23 and in mesoporous silica.70,71 The complete lack of endothermic peaks at this degree of loading also indicates that only an insignificant, if any, amount of ITZ resides on the outer surface of the carrier particles. The outer surface area of the carrier particles was negligible compared to the internal surface area and only has a limited ability to interact with the ITZ in order to suppresses the crystallisation of the substance.26 When the drug loading level increased, crystallisation was no longer completely suppressed, irrespectively to the sample identity (ESI, Fig. S22 and Table S4†). Whereas samples E (pore size ∼ 5.1 nm) and A (pore size ∼ 13 nm) were able to completely stabilise ITZ in the amorphous state at a drug loading degree of 45%, a weak and broad endothermic peak located at the melting point temperature of crystalline ITZ was detected for sample D (pore size ∼ 20 nm). Similar humps were observed for all samples at drug loading degrees of 60%. This illustrated that large pore sizes offered more space for the incorporated drug molecules to rearrange and crystallise. Furthermore, the finding also emphasised that for optimised stabilisation effect of the drug, the pore size of the drug carrier must correlate to the size of the drug. The samples with ∼5.1 and 13 nm average pores size could host relatively large amounts of ITZ (having the rectangular cuboid dimensions of 2.97 nm × 0.93 nm × 0.69 nm)72 and successfully hindered the crystallisation of ITZ (low solubility form of the substance). Since ITZ remained amorphous in all samples at a loading degree of 30%, this degree of drug loading was used for the drug release measurements.
The time-resolved release of ITZ from the grounded transparent carrier particles into simulated gastric fluid (pH ∼ 1.3) is shown in Fig. 6. As observed in the figure, the release rate during the first 30 min could be enhanced with a factor of ∼23 (release from sample D as compared to pure ITZ) when ITZ was loaded into transparent MMC. The corresponding release rate enhancement for samples A and E were ∼17 and ∼13, respectively.
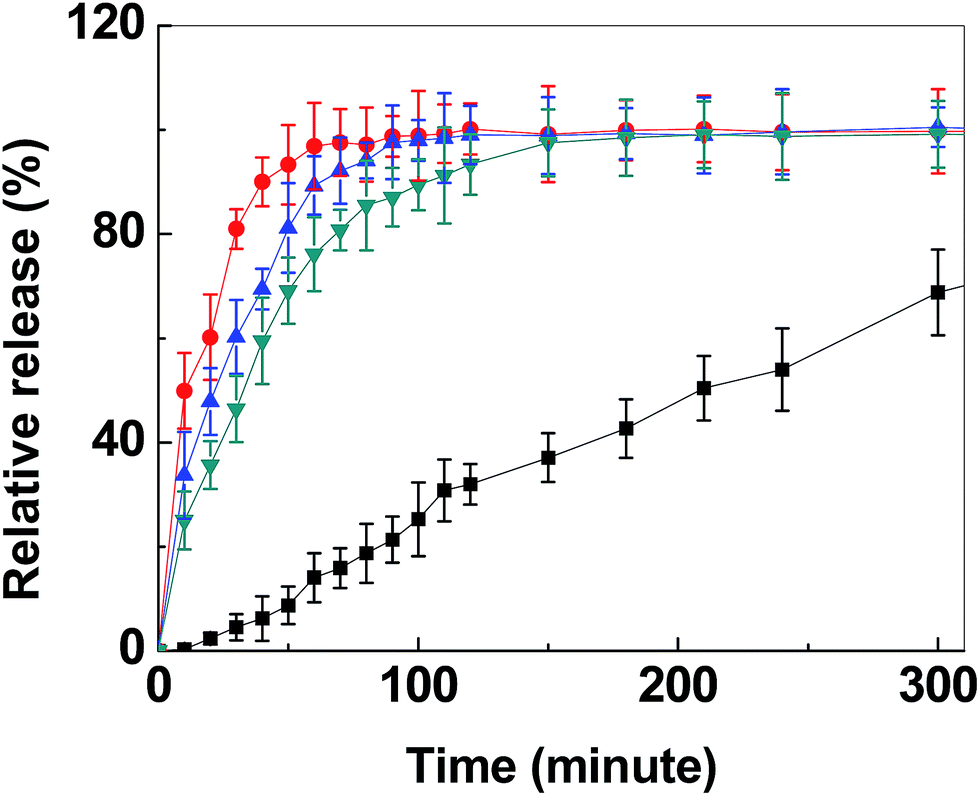 | ||
| Fig. 6 Time resolved release in simulated gastric fluid of itraconazole (ITZ) loaded into transparent MMC powder with average pore sizes of 20 nm (red curve), 13 nm (blue curve) and 5.1 nm (green curve) as well as dissolution of the pure crystalline drug (black curve). The lines are drawn as guides for the eye. The error bars signify variations over three measurements. | ||
During the first 30 min of release, no visible changes to the powder in the dissolution vessel were observed. The time required for the full release of ITZ also differed between transparent MMC drug carriers with different pore sizes. The large pore (∼20 nm) carrier was able to fully release the loaded drug molecules within 60 min. This was noticeably faster than its smaller pore size counterparts and the pure crystalline form of the drug, which was anticipated since a larger average pore size inevitably enhances the constrictivity to tortuosity ratio and, hence, the effective diffusion coefficient of the drug from the carrier to the dissolution medium.23,73
Several hours after the initial release, powder dissolution was observed. This was expected due to the acidic conditions in the release medium.
The results showed clearly that the amorphous phase stabilisation properties, as well as the release rate, could be tuned by adjusting the pore size of the carrier. This finding opens up for new possibilities to stabilise the vast number of amorphous compounds in the R&D pipe-line of big pharmaceutical companies.74 In addition, enteric coating of the drug loaded transparent MMC powder may be employed to further optimise the release rate. Enteric coating will hinder the release of the drug and particle dissolution in the acidic environment of the stomach. The drug release process can then be delayed until the coated particles have reached the intestines. It should be noted that both MgCO3 and MgO are GRAS (Generally Recognized As Safe) listed by the U.S. Food and Drug Administration and have E-numbers (E504 for MgCO3 and E530 for MgO).75
Conclusion
In summary, we have revealed the details of the nanocomposite structure of a mesoporous magnesium carbonate (MMC). We also showed how the pore size of the material can be adjusted between ∼3 and ∼20 nm without the addition of organic templates or swelling agents. Pore size control was achieved by controlling the energy input (temperature, stirring etc.) during the gel drying/powder formation step of the synthesis. The material was found to form from the hierarchical aggregation of MgO nanocrystals and MgCO3 nanoparticles that are approximately 2–5 nm in size. These 2–5 nm nanoparticles first form ∼30 nm nanoparticles, then these ∼30 nm nanoparticles aggregate to form MMC particles. The resulting aggregates are transparent powders with high porosity. By tuning the pore structure of the material it was found to be possible to tailor the amorphous phase stabilisation properties exerted on the poorly soluble antifungal drug itraconazole. The drug release rate from the material could also be tailored and the drug release was diffusion/pore size controlled.Acknowledgements
M. S. (Uppsala University) thanks the Swedish Research Council (grant # 2014-3929) and the Swedish Energy Agency (grant # P38273-1) for financial support. X. Z. (Stockholm University) thanks the Knut and Alice Wallenberg Foundation for the financial supports of the 3DEM-NATUR project and the TEMs. P. Z. thanks the China Scholarship Council (CSC) for financial support of the present work. Wei Wan is acknowledged for the support in electron tomography. Simon Gustafsson is acknowledged for valuable help regarding centrifugation and temperature control of the pore size. M. S. (PANalytical B.V.) acknowledges Milen Gateshki and Peter Munk for useful discussions on PDF measurements. Olga Narygina is acknowledged for help with PXRD measurements.References
- K. E. Drexler, Engines of Creation: The Coming Era of Nanotechnology, Anchor Books, New York, 1986 Search PubMed.
- O. Cheung, Z. Bacsik, P. Krokidas, A. Mace, A. Laaksonen and N. Hedin, Langmuir, 2014, 30, 9682–9690 CrossRef CAS PubMed.
- O. Cheung, Z. Bacsik, Q. Liu, A. Mace and N. Hedin, Appl. Energy, 2013, 112, 1326–1336 CrossRef CAS.
- O. Cheung, D. Wardecki, Z. Bacsik, P. Vasiliev, L. B. McCusker and N. Hedin, Phys. Chem. Chem. Phys., 2016, 18, 16080–16083 RSC.
- O. Cheung, J. Su, Z. Bacsik, J. Li, L. Samain, X. Zou and N. Hedin, Microporous Mesoporous Mater., 2014, 198, 63–73 CrossRef CAS.
- S. Ferdov, Langmuir, 2010, 26, 2684–2687 CrossRef CAS PubMed.
- R. Hutter, T. Mallat, D. Dutoit and A. Baiker, Top. Catal., 1996, 3, 421–436 CrossRef CAS.
- J. M. Campos, J. P. Lourenço, H. Cramail and M. R. Ribeiro, Prog. Polym. Sci., 2012, 37, 1764–1804 CrossRef CAS.
- J. Galán, A. Rodríguez, J. M. Gómez, S. J. Allen and G. M. Walker, Chem. Eng. J., 2013, 219, 62–68 CrossRef.
- X. Peng, D. Huang, T. Odoom-Wubah, D. Fu, J. Huang and Q. Qin, J. Colloid Interface Sci., 2014, 430, 272–282 CrossRef CAS PubMed.
- Y. Dong, H. Lin, Q. Jin, L. Li, D. Wang, D. Zhou and F. Qu, J. Mater. Chem. A, 2013, 1, 7391–7398 CAS.
- F. Tang, L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS PubMed.
- Z. Alothman, Materials, 2012, 5, 2874 CrossRef CAS.
- T. Asefa and Z. Tao, Can. J. Chem., 2012, 90, 1015–1031 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T.-W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. A. Schlenker, J. Am. Chem. Soc., 1992, 27, 10834–10843 CrossRef.
- S. Y. Chang and T. A. Ring, J. Non-Cryst. Solids, 1992, 147–148, 56–61 CrossRef CAS.
- C.-Y. Chen, S. L. Burkett, H.-X. Li and M. E. Davis, Microporous Mater., 1993, 2, 27–34 CrossRef CAS.
- A. Firouzi, D. Kumar, L. M. Bull, T. Besier, P. Sieger, Q. Huo, S. A. Walker, J. A. Zasadzinski, C. Glinka and J. Nicol, et al. , Science, 1995, 267, 1138–1143 CrossRef CAS PubMed.
- Q. Huo, D. I. Margolese, U. Ciesla, P. Feng, T. E. Gier, P. Sieger, R. Leon, P. M. Petroff, F. Schuth and G. D. Stucky, Nature, 1994, 368, 317–321 CrossRef CAS.
- C. Boissière, M. Kümmel, M. Persin, A. Larbot and E. Prouzet, Adv. Funct. Mater., 2001, 11, 129–135 CrossRef.
- U. Brohede, R. Atluri, A. E. Garcia-Bennett and M. Strømme, Curr. Drug Delivery, 2008, 5, 177–185 CrossRef CAS.
- P. Zhang, J. Forsgren and M. Strømme, Int. J. Pharm., 2014, 472, 185–191 CrossRef CAS PubMed.
- P. Zhang, T. Z. Gómez De La Torre, J. Forsgren, C. A. S. Bergström and M. Strømme, J. Pharm. Sci., 2016, 105, 657 CrossRef CAS PubMed.
- M. Strømme, U. Brohede, R. Atluri and A. E. Garcia-Bennett, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2009, 1, 140–148 CrossRef PubMed.
- X. Xia, C. Zhou, L. Ballell and A. E. Garcia-Bennett, ChemMedChem, 2012, 7, 43–48 CrossRef CAS PubMed.
- J. Forsgren, M. Andersson, P. Nilsson and A. Mihranyan, Adv. Healthcare Mater., 2013, 2, 1469–1476 CrossRef CAS PubMed.
- M. Colilla, A. Baeza and M. Vallet-Regí, in The Sol–Gel Handbook, Wiley-VCH Verlag GmbH & Co. KGaA, 2015, pp. 1309–1344 Search PubMed.
- B. Fadeel, B. Kasemo, M. Malmsten and M. Strømme, J. Intern. Med., 2010, 267, 2–8 CrossRef CAS PubMed.
- M. Vallet-Regí, J. Intern. Med., 2010, 267, 22–43 CrossRef PubMed.
- H. Vallhov, N. Kupferschmidt, S. Gabrielsson, S. Paulie, M. Strømme, A. E. Garcia-Bennett and A. Scheynius, Small, 2012, 8, 2116–2124 CrossRef CAS PubMed.
- H. Vallhov, S. Gabrielsson, M. Strømme, A. Scheynius and A. E. Garcia-Bennett, Nano Lett., 2007, 7, 3576–3582 CrossRef CAS PubMed.
- F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 3216–3251 CrossRef CAS PubMed.
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS PubMed.
- A. Corma, Chem. Rev., 1997, 97, 2373–2420 CrossRef CAS PubMed.
- A. Sayari, Chem. Mater., 1996, 8, 1840–1852 CrossRef CAS.
- J. Y. Ying, C. P. Mehnert and M. S. Wong, Angew. Chem., Int. Ed., 1999, 38, 56–77 CrossRef CAS.
- Z. Bacsik, R. Atluri, A. E. Garcia-Bennett and N. Hedin, Langmuir, 2010, 26, 10013–10024 CrossRef CAS PubMed.
- S.-H. Wu, C.-Y. Mou and H.-P. Lin, Chem. Soc. Rev., 2013, 42, 3862–3875 RSC.
- T. Yamada, H. Zhou, K. Asai and I. Honma, Mater. Lett., 2002, 56, 93–96 CrossRef CAS.
- M. Widenmeyer and R. Anwander, Chem. Mater., 2002, 14, 1827–1831 CrossRef CAS.
- M. Kruk, Acc. Chem. Res., 2012, 45, 1678–1687 CrossRef CAS PubMed.
- D. Niu, Z. Liu, Y. Li, X. Luo, J. Zhang, J. Gong and J. Shi, Adv. Mater., 2014, 26, 4947–4953 CrossRef CAS PubMed.
- D. K. Singh, B. V. V. S. Pavan Kumar and M. Eswaramoorthy, Nanoscale, 2015, 7, 13358–13362 RSC.
- G. Song, X. Zhu, R. Chen, Q. Liao, Y.-D. Ding and L. Chen, RSC Adv., 2016, 6, 19069–19077 RSC.
- L. Zhang, X. Jing, R. Li, Q. Liu, J. Liu, H. Zhang, S. Hu and J. Wang, RSC Adv., 2015, 5, 23144–23151 RSC.
- J. G. Vitillo, RSC Adv., 2015, 5, 36192–36239 RSC.
- J. Forsgren, S. Frykstrand, K. Grandfield, A. Mihranyan and M. Strømme, PLoS One, 2013, 8, e68486 CAS.
- S. Frykstrand, J. Forsgren, A. Mihranyan and M. Strømme, Microporous Mesoporous Mater., 2014, 190, 99–104 CrossRef CAS.
- I. Pochard, S. Frykstrand, O. Ahlström, J. Forsgren and M. Strømme, J. Appl. Phys., 2014, 115, 044306–044310 CrossRef.
- I. Pochard, S. Frykstrand, J. Eriksson, S. Gustafsson, K. Welch and M. Strømme, J. Phys. Chem. C, 2015, 119, 15680–15688 CAS.
- S. Frykstrand, J. Forsgren, P. Zhang, M. Strømme and N. Ferraz, J. Biomater. Nanobiotechnol., 2015, 6, 257 CrossRef.
- S. Frykstrand, J. Forsgren, O. Cheung, P. Zhang, J. Hong, M. Stromme and N. Ferraz, RSC Adv., 2016, 6, 52810–52816 RSC.
- S. Ardizzone, C. Bianchi, M. Fadoni and B. Vercelli, Appl. Surf. Sci., 1997, 119, 253–259 CrossRef CAS.
- A. Reiss Celeste, A. Kharchenko and M. Gateshki, Z. Kristallogr., 2012, 257 CrossRef.
- G. Confalonieri, M. Dapiaggi, M. Sommariva, M. Gateshki, A. N. Fitch and A. Bernasconi, Powder Diffr., 2015, 30, S65–S69 CrossRef CAS.
- M. Sommariva, M. Gateshki, J.-A. Gertenbach, J. Bolze, U. König, B. Ş. Vasile and V.-A. Surdu, Powder Diffr., 2014, 29, S47–S53 CrossRef CAS.
- T. Degen, M. Sadki, E. Bron, U. König and G. Nénert, Powder Diffr., 2014, 29, S13–S18 CrossRef CAS.
- C. L. Farrow, P. Juhas, J. W. Liu, D. Bryndin, E. S. Božin, J. Bloch, T. Proffen and S. J. L. Billinge, J. Phys.: Condens. Matter, 2007, 19, 335219 CrossRef CAS PubMed.
- F. Amat, F. Moussavi, L. R. Comolli, G. Elidan, K. H. Downing and M. Horowitz, J. Struct. Biol., 2008, 161, 260–275 CrossRef PubMed.
- J. R. Kremer, D. N. Mastronarde and J. R. McIntosh, J. Struct. Biol., 1996, 116, 71–76 CrossRef CAS PubMed.
- D. N. Mastronarde, J. Struct. Biol., 1997, 120, 343–352 CrossRef CAS PubMed.
- S. Frykstrand, C. Strietzel, J. Forsgren, J. Angström, V. Potin and M. Strømme, CrystEngComm, 2014, 16, 10837–10844 RSC.
- S. Gryglewicz, Appl. Catal., A, 2000, 192, 23–28 CrossRef CAS.
- F. Daniel, F. Flieder and F. LeClerc, Restaurator, 1990, 11, 179–207 CrossRef CAS.
- L. Green and M. Leese, Restaurator, 1991, 12, 147–162 CAS.
- R. M. Dell and S. W. Weller, Trans. Faraday Soc., 1959, 55, 2203–2220 RSC.
- T. Kornprobst and J. Plank, J. Non-Cryst. Solids, 2013, 361, 100–105 CrossRef CAS.
- M. Lee, E. Y. Lee, D. Lee and B. J. Park, Soft Matter, 2015, 11, 2067–2079 RSC.
- A. Glasmacher and A. Prentice, Clin. Microbiol. Infect., 2006, 12, 84–90 CrossRef CAS.
- E. Verraedt, G. Van den Mooter and J. A. Martens, J. Pharm. Sci., 2011, 100, 4295–4301 CrossRef CAS PubMed.
- S. Roy Chowdhury, A. M. Peters, D. H. A. Blank and J. E. Elshof, J. Membr. Sci., 2006, 279, 276–281 CrossRef.
- R. Mellaerts, J. A. G. Jammaer, M. Van Speybroeck, H. Chen, J. V. Humbeeck, P. Augustijns, G. Van den Mooter and J. A. Martens, Langmuir, 2008, 24, 8651–8659 CrossRef CAS PubMed.
- F. A. Coutelieris and J. M. P. Q. Delgado, Transport Processes in Porous Media, Springer-Verlag Berlin Heidelberg, New York, 2012 Search PubMed.
- R. Laitinen, K. Löbmann, C. J. Strachan, H. Grohganz and T. Rades, Int. J. Pharm., 2013, 453, 65–79 CrossRef CAS PubMed.
- U.S. Food and Drug Administration-FDA, Food Additive Status List, U.S. Food and Drug Administration, 2015 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra14171d |
| This journal is © The Royal Society of Chemistry 2016 |