
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe design of liquid crystalline bistolane-based materials with extremely high birefringence†
Yuki
Arakawa
ab,
Sungmin
Kang
*a,
Hideto
Tsuji
b,
Junji
Watanabe
a and
Gen-ichi
Konishi
*a
aDepartment of Organic and Polymeric Materials, Tokyo Institute of Technology, O-okayama, Meguro-ku, Tokyo 152-8552, Japan. E-mail: skang@polymer.titech.ac.jp; konishi.g.aa@m.titech.ac.jp
bDepartment of Environmental and Life Sciences, Graduate School of Engineering, Toyohashi University of Technology, Tempaku-cho, Toyohashi, Aichi 441-8580, Japan
First published on 22nd September 2016
Abstract
We designed three new bistolanes with alkylsulfanyl groups. A symmetric derivative contains short terminal methylsulfanyl groups (–SCH3) on both ends, while two asymmetric derivatives contain a methylsulfanyl group on one end, as well as a highly polarizable cyano (–CN) or isothiocyanate (–NCS) group on the other end. These materials exhibited a well-defined enantiotropic nematic phase, as well as an extremely high birefringence (Δn > 0.6). Most notably, the NCS derivative achieves an extremely high value of 0.77 at 550 nm. These results should be helpful for the design and synthesis of novel nematic materials with high birefringence.
Introduction
Over the last two decades, liquid crystals (LCs) have attracted substantial attention as materials with high birefringence (Δn). Their intriguing features include self-alignment, anisotropy, and sensitivity to external fields, which provides access to a range of optical and electric materials, as well as display films.1 So far, these materials have been used extensively as cholesteric (Ch) films,1c,2 including films for selective and wide reflection, laser emission, holographic applications,3 and LC lenses.4 Most high-birefringence LC materials exhibit Δn values of 0.2–0.6. However, in order to improve their performance in the aforementioned applications, values beyond this range are required, as well as thinner LC films and a wider reflection band for circularly polarized light from Ch films. To the best of our knowledge, reports on such materials with Δn > 0.6 still remain extremely scarce.5The typical design for high-Δn LC materials includes the fabrication of rod-like cores via a combination of aromatic rings and/or unsaturated bonds.3,5,6 High molecular polarizability anisotropies are usually attained by attaching highly polarizable substituents, e.g. cyano (–CN) and isothiocyanato (–NCS) groups, to the terminal positions of these compounds.6
Recently, we described novel alkylsulfanyl bistolane-based rod-like LC materials, whose mesophases are induced by introducing a fluorine atom at the lateral position of the central benzene ring.7 Owing to the S⋯S interactions and high polarizability of the sulphur atom, these materials exhibit remarkable characteristics, including high Δn and high order parameters. However, various performance-limiting issues remain unresolved. For example, the mesophase range of these materials is narrower compared to that of other derivatives that contain carbon or oxygen atoms.5a,7,8 Moreover, their Δn values are still lower than those of extremely high-Δn materials, the so-called super-high-Δn (SHB) materials.5b
In this study, we present novel bistolane-based LC materials with extremely high Δn (Δn > 0.7). In order to achieve such extremely high Δn, we aimed to increase the mesophase range and the polarizability anisotropy. Therefore, we designed three bistolane-based LC compounds (Fig. 1): two asymmetric molecules, containing a methylsulfanyl group at one of the terminal positions and a highly polarizable isothiocyanato (1) or cyano group (2) at the other, as well as a symmetric methylsulfanyl derivative (3) with enhanced intermolecular S⋯S interactions. The present molecular design uses methylsulfanyl groups, i.e. the shortest possible alkyl chain, to realize extremely high Δn. Relative to longer alkyl chains, shorter ones should not only lead to a higher density of the mesogenic moiety in direction of the long molecular axis to provide a high extraordinary refractive index, but also to a wider nematic (N) phase for calamitic LC molecules.
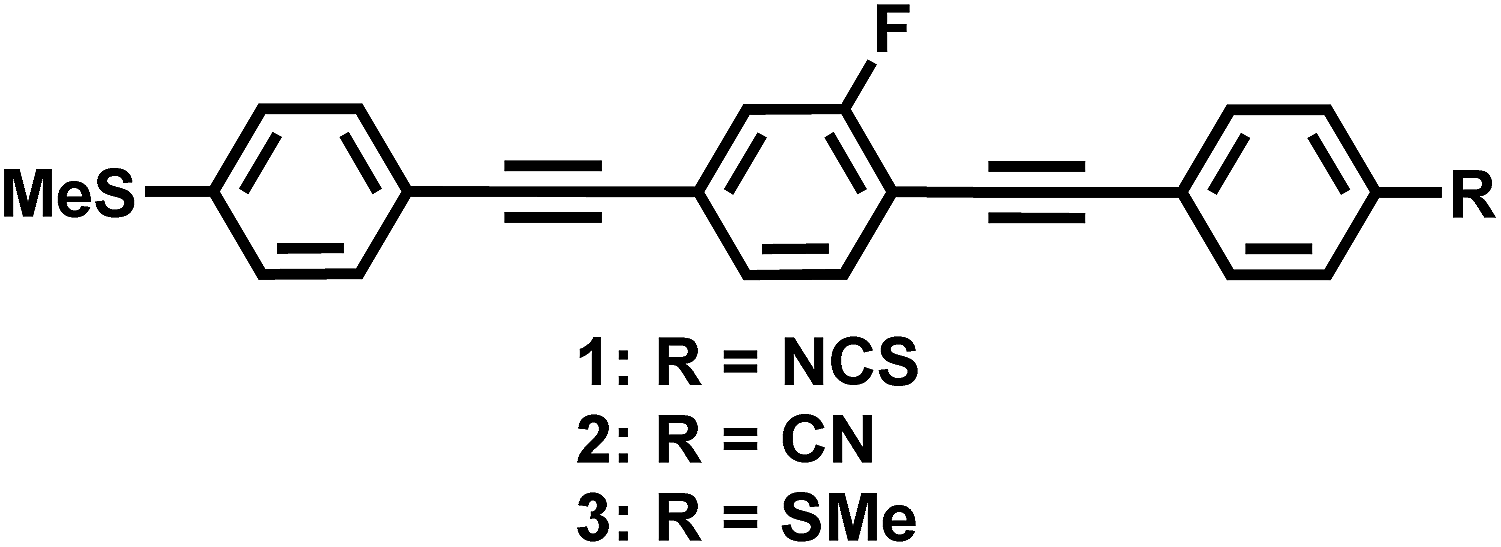 | ||
| Fig. 1 Chemical structures of 1–3. | ||
Experimental
Instruments
1H and 13C NMR spectra were measured in CDCl3 on JEOL LNM-EX 400 or Bruker DPX300S spectrometers at room temperature, using tetramethylsilane (TMS) as the internal standard. Liquid crystalline textures were examined by polarizing optical microscopy (POM) on a Leica DM2500P microscopy with a Mettler FP90 hot stage. Transition temperatures and enthalpy changes were measured by differential scanning calorimetry (DSC) on a Perkin Elmer DSC7, whereby heating and cooling scans were carried out using a temperature gradient of 10 °C min−1. Measurements of extraordinary (ne) and ordinary (no) refractive indices and of Δn were carried out in uniaxially aligned nematic cells, which contained an indium tin oxide (ITO) layer and were purchased from EHC. The cell gaps (d) were 2 or 3 μm. The transmittance of light under crossed nicol conditions was observed as a function of wavelength by a microscopic/spectroscopic method using a Nikon LV100 Pol optical microscope equipped with a USB4000 (Ocean photonics) spectrometer.Synthesis
Chemicals were obtained from TCI, Wako, or Nacalai Tesque (Japan) and used as received. The synthetic procedures for all compounds are described and the synthetic routes are shown in Schemes 1–3. The characterization data for 1–3 include 1H NMR spectra, as well as high-resolution mass spectrometry (HRMS) data.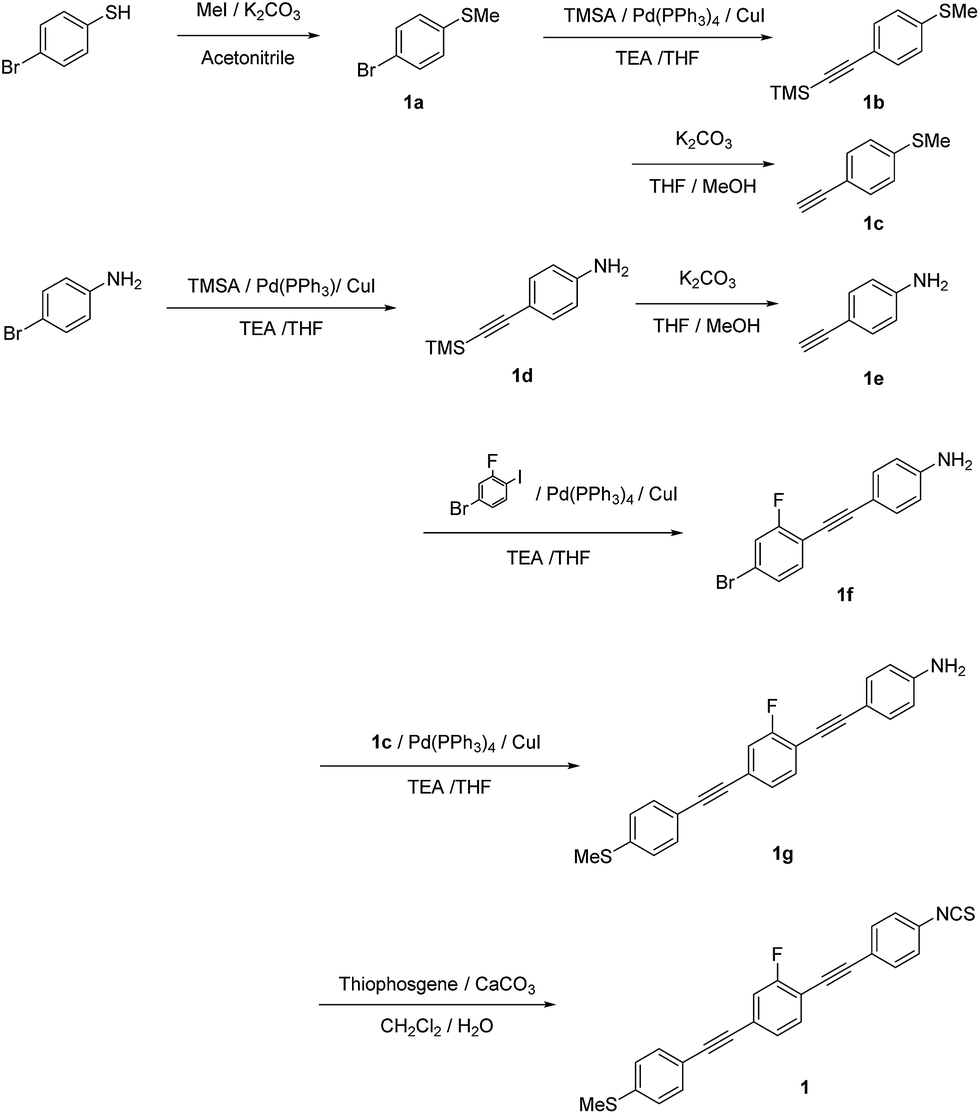 | ||
| Scheme 1 Synthetic route to 1. | ||
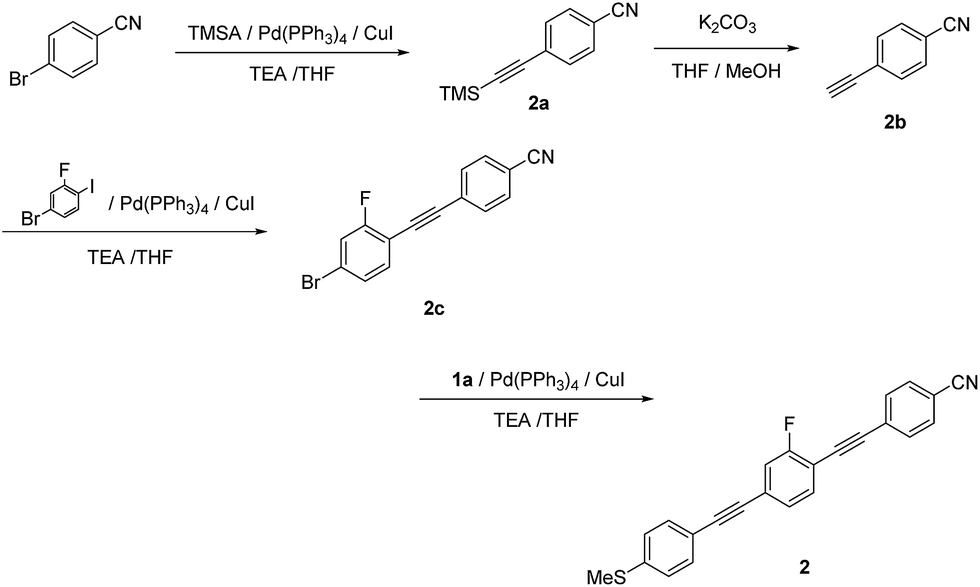 | ||
| Scheme 2 Synthetic route to 2. | ||
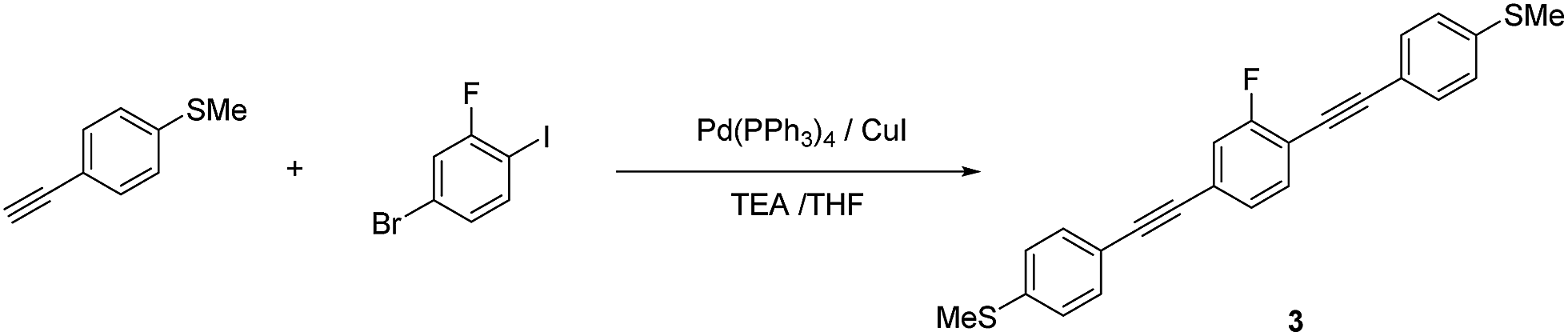 | ||
| Scheme 3 Synthetic route to 3. | ||
1-Bromo-4-(methylthio)benzene (1a). A mixture of 4-bromobenzenethiol (7.00 g, 37.0 mmol), iodomethane (MeI; 4.61 mL, 74.0 mmol), K2CO3 (10.2 g, 74.0 mmol), and MeCN (60 mL) was heated to reflux for 12 h. The reaction mixture was cooled to room temperature, extracted with ethyl acetate, washed with water and brine, and dried over MgSO4. After removing the solvent under reduced pressure, 1a was obtained as a colorless solid (7.41 g, 99%), which was used without further purification. 1H NMR (400 MHz, CDCl3) δ 7.39 (d, J = 8.8 Hz, Ar-H, 2H), 7.12 (d, J = 8.8 Hz, Ar-H, 2H), 2.47 (s, Ar-S–CH3, 3H) ppm.
1-Methylthio-4-[2-(trimethylsilyl)ethynyl]benzene (1b) (general procedure for Sonogashira coupling). A mixture of 1a (7.41 g, 36.5 mmol), Pd(PPh3)4 (0.42 g, 0.37 mmol), CuI (70.0 mg, 0.37 mmol), trimethylsilylacetylene (TMSA; 7.6 mL, 54.7 mmol), triethylamine (TEA; 40 mL), and tetrahydrofuran (THF; 40 mL; degassed by argon sparging) was stirred at 45 °C for 24 h. After cooling to room temperature, the reaction mixture was filtered, extracted with ethyl acetate, washed with aqueous HCl (2 M) and brine, and dried over MgSO4. After evaporating the solvent, the residue was purified by column chromatography on silica gel (eluent: ethyl acetate/hexane = 1/10, v/v) to afford a pale brown oil (7.58 g, 94%). 1H NMR (400 MHz, CDCl3) δ 7.37 (d, J = 8.8 Hz, Ar-H, 2H), 7.15 (d, J = 8.8 Hz, Ar-H, 2H), 2.47 (s, Ar-S–CH3, 3H), 0.25 (s, Si–(CH3)3, 9H) ppm.
1-Ethynyl-4-methylthiobenzene (1c). A mixture of 1b (3.50 g, 15.9 mmol), K2CO3 (4.40 g, 31.8 mmol), MeOH (20 mL), and THF (20 mL) was stirred at room temperature for 30 min. Then, the reaction mixture was extracted with ethyl acetate and washed with brine. After evaporating the solvent, 1c was obtained as a colorless solid (0.72 g, >99%). 1H NMR (400 MHz, CDCl3) δ 7.40 (d, J = 8.4 Hz, Ar-H, 2H), 7.17 (d, J = 8.4 Hz, Ar-H, 2H), 3.07 (s, C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H, 1H), 2.48 (s, Ar-S–CH3, 3H) ppm.
C–H, 1H), 2.48 (s, Ar-S–CH3, 3H) ppm.
4-[2-(Trimethylsilyl)ethynyl]aniline (1d). Compound 1d was synthesized according to the generic Sonogashira coupling conditions outlined for the preparation of 1b: 4-iodoaniline (8.00 g, 36.5 mmol), trimethylsilylacetylene (7.58 mL, 54.8 mmol), TEA (40 mL), THF (40 mL), Pd(PPh3)4 (0.420 g, 0.365 mmol), and CuI (69.5 mg, 0.365 mmol). Purification by column chromatography on silica gel (eluent: ethyl acetate/hexane = 1/3, v/v) afforded 1d as a pale brown solid in 77% yield. 1H NMR (300 MHz, CDCl3) δ 7.23 (d, J = 8.4 Hz, Ar-H, 2H), 6.57 (d, J = 8.4 Hz, Ar-H, 2H), 3.79 (brs, –NH2, 2H), 0.23 (s, Si–(CH3)3, 9H) ppm.
4-Ethynylaniline (1e). Compound 1e was synthesized according to the method for the deprotection of trimethylsilyl groups used for the preparation of 1c: 1d (5.27 g, 27.8 mmol), K2CO3 (5.77 g, 41.8 mmol), MeOH (30 mL), and THF (30 mL). Yield: 99%. Pale brown solid. 1H NMR (300 MHz, CDCl3) δ 7.30 (d, J = 8.4 Hz, Ar-H, 2H), 6.60 (d, J = 8.4 Hz, Ar-H, 2H), 3.82 (brs, –NH2, 2H), 2.96 (s, C
C–H, 1H) ppm.
4-[2-(4-Bromo-2-fluorophenyl)ethynyl]aniline (1f). Compound 1f was synthesized according to the generic Sonogashira coupling conditions outlined for the preparation of 1b: 1e (3.26 g, 27.8 mmol), 4-bromo-2-fluoroiodobenzene (10.0 g, 33.4 mmol), TEA (40 mL), THF (40 mL), Pd(PPh3)4 (0.320 g, 0.280 mmol), and CuI (53.0 mg, 0.280 mmol). Purification by column chromatography on silica gel (eluent: ethyl acetate/hexane = 1/3, v/v) afforded 1f as a colorless solid in 77% yield. 1H NMR (300 MHz, CDCl3) δ 7.35 (d, J = 8.1 Hz, Ar-H, 2H), 7.33-7.22 (m, Ar-H, 3H), 6.64 (d, J = 8.1 Hz, Ar-H, 2H), 3.87 (brs, –NH2, 2H) ppm.
2-Fluoro-1-[2-(4-aminophenyl)ethynyl]-4-[2-(4-thiophenyl)ethynyl]benzene (1g). Compound 1g was synthesized according to the generic Sonogashira coupling conditions outlined for the preparation of 1b: 1c (0.34 g 2.27 mmol), 1f (0.66 g, 2.27 mmol), TEA (5 mL), THF (5 mL), Pd(PPh3)4 (0.13 g, 0.11 mmol), and CuI (21 mg, 0.11 mmol). Purification by column chromatography on silica gel (eluent: chloroform/hexane = 1/2, v/v) afforded 1g as a pale yellow solid in 27% yield. 1H NMR (300 MHz, CDCl3) δ 7.46–40 (m, Ar-H, 3H), 7.36 (d, J = 8.4 Hz, Ar-H, 2H), 7.25–7.17 (m, Ar-H, 4H), 6.65 (d, J = 8.1 Hz, Ar-H, 2H), 3.86 (brs, –NH2, 2H), 2.50 (s, –CH3, 3H) ppm.
2-Fluoro-1-[2-(4-isothiocyanatophenyl)ethynyl]-4-[2-(4-methylthiophenyl)ethynyl]benzene (1). A mixture of 1g (0.650 g, 1.82 mmol), CaCO3 (0.360 g, 3.64 mmol), dichloromethane (25 mL), and water (10 mL) was cooled to 0 °C. Then, thiophosgene (0.19 mL, 2.52 mmol) was added dropwise, and the resulting mixture was stirred for 12 h at room temperature. Subsequently, the mixture was extracted with CHCl3, washed with water and brine, and dried over MgSO4. After removing all volatiles, the residue was purified by column chromatography on silica gel (eluent: chloroform/hexane = 1/2, v/v). The residue was recrystallized from CHCl3 and hexane to afford the target compound as a pale brown solid in 77%. 1H NMR (300 MHz, CDCl3) δ 7.55 (d, J = 8.4 Hz, Ar-H, 2H), 7.51–7.41 (m, Ar-H, 3H), 7.30 (d, J = 8.4 Hz, Ar-H, 2H), 7.24 (d, J = 8.4 Hz, Ar-H, 4H), 2.53 (s, –CH3, 3H) ppm. HRMS-FAB+ (m/z): [M]+ calcd for C24H14FNS2, 399.0552; found, 399.0556.
1-Cyano-4-[2-(trimethylsilyl)ethynyl]benzene (2a). Compound 2a was synthesized according to the general Sonogashira coupling conditions outlined for the preparation of 1b: 4-bromobenzonitrile (5.00 g, 27.5 mmol), trimethylsilylacetylene (5.70 mL, 41.2 mmol), TEA (20 mL), THF (20 mL), Pd(PPh3)4 (0.320 g, 0.275 mmol), and CuI (52.3 mg, 0.275 mmol); purification by column chromatography on silica gel (eluent: CHCl3/hexane = 1/6, v/v); yield: >99%; pale yellow solid. 1H NMR (300 MHz, CDCl3) δ 7.59 (d, J = 8.4 Hz, Ar-H, 2H), 7.53 (d, J = 8.4 Hz, Ar-H, 2H), 3.07 (s, C
C–H, 1H), 0.28 (s, Si–(CH3)3, 9H) ppm.
1-Cyano-4-ethynylbenzene (2b). Compound 2b was synthesized according to the general method for the deprotection of trimethylsilyl groups used for the preparation of 1c: 2a (5.48 g, 27.5 mmol), K2CO3 (11.4 g, 82.5 mmol), MeOH (40 mL), and THF (40 mL); yield: 54%; colorless solid. 1H NMR (300 MHz, CDCl3) δ 7.62 (d, J = 8.4 Hz, Ar-H, 2H), 7.57 (d, J = 8.4 Hz, Ar-H, 2H), 3.30 (s, C
C–H, 1H) ppm.
4-[2-(4-Bromo-2-fluorophenyl)ethynyl]benzonitrile (2c). Compound 2c was synthesized according to the general Sonogashira coupling conditions used for the preparation of 1b: 4-ethynylbenzonitrile (0.900 g, 7.08 mmol), 4-bromo-2-fluoroiodobenzene (2.55 g, 8.49 mmol), TEA (15 mL), THF (15 mL), Pd(PPh3)4 (0.245 g, 0.212 mmol), and CuI (40.4 mg, 0.212 mmol); purification by column chromatography on silica gel (eluent: CHCl3/hexane = 1/3, v/v); yield: 86%; colorless solid. 1H NMR (300 MHz, CDCl3) δ 7.66 (d, J = 8.7 Hz, Ar-H, 2H), 7.62 (d, J = 8.7 Hz, Ar-H, 2H), 7.42–7.29 (m, Ar-H, 3H) ppm, 13C NMR (300 MHz) δ 86.4, 94.0 (d, J = 3.7 Hz), 110.5, 110.7, 112.5, 118.8, 119.8, 120.1, 124.1 (d, J = 9.0 Hz), 127.8, 128.1 (d, J = 3.7 Hz), 132.5 (d, J = 3.0 Hz), 132.5 (d, J = 3.0 Hz), 134.6 (d, J = 1.5 Hz), 161.1, 164.5 ppm.
2-Fluoro-1-[2-(4-cyanophenyl)ethynyl]-4-[2-(4-methylthiophenyl)ethynyl]benzene (2). Compound 2 was synthesized according to the general Sonogashira coupling conditions used for the preparation of 1b: 2c (0.100 g, 0.330 mmol), 1c (53.0 mg, 0.360 mmol), TEA (5 mL), THF (5 mL), Pd(PPh3)4 (19.0 mg, 17.0 μmol), and CuI (3.10 mg, 17.0 μmol); purification by column chromatography on silica gel (eluent: CHCl3/hexane = 1/2, v/v), followed by recrystallisation from CHCl3/hexane; yield: 82%; colorless solid; 1H NMR (400 MHz, CDCl3) δ 7.66 (d, J = 8.4 Hz, Ar-H, 2H), 7.63 (d, J = 8.8 Hz, Ar-H, 2H), 7.48 (s, Ar-H, 1H), 7.44 (d, J = 8.8 Hz, Ar-H, 2H), 7.30 (d, J = 9.6 Hz, Ar-H, 1H), 7.29 (d, J = 9.6 Hz, Ar-H, 1H), 7.22 (d, J = 8.4 Hz, Ar-H, 2H), 2.51 (s, –CH3, 3H) ppm. HRMS-FAB+ (m/z): [M]+ calcd for C24H14FNS, 367.0831; found, 367.0822.
1,4-Bis[4-(methylsulfanyl)phenyl]ethynyl-2-fluorobenzene (3). Compound 3 was synthesized according to the general Sonogashira coupling conditions outlined for the preparation of 1b: 1c (0.74 mmol, 0.11 g), 1-bromo-3-fluoro-4-iodobenzene (0.37 mmol, 0.11 g), Pd(PPh3)4 (37 μmol, 43 mg), CuI (37 μmol, 43 mg), TEA (5 mL), and THF (5 mL); purification by column chromatography on silica gel (eluent: CHCl3/hexane = 1/2, v/v), followed by recrystallisation from CHCl3/hexane; colorless solid (75.3 mg, 52%). 1H NMR (400 MHz, CDCl3) δ 7.49–7.42 (m, Ar-H, 5H), 7.25–7.20 (m, Ar-H, 6H), 2.51 (s, –CH3, 6H) ppm. HRMS-FAB+ (m/z): [M]+ calcd for C24H17FNS2, 388.0756; found, 388.0746.
Results and discussion
The phase transition temperatures for 1–3, determined by differential scanning calorimetry and polarized optical microscopy are summarized in Table 1, and the corresponding DSC curves are shown in Fig. S1–S3.† As shown in Table 1, 1–3 exhibit solely enantiotropic N phases without further higher-ordered smectic phases; the marble and schlieren textures indicative of N phases are shown in Fig. S4–S6.† Here, it is important to note that asymmetrically substituted 1 and 2 exhibit significantly wider N phases than symmetric 3. Relative to the corresponding hexylsulfanyl-derivative, 3 shows a wider mesophase.7 The melting temperatures (Tm), indicating the transition from the crystal (Cr) to the N phase, of 1 (149.9 °C) and 2 (179.0 °C) are considerably lower than that of 3 (203.8 °C). Conversely, the clearing temperatures (Ti) for 1 and 2 are higher than that for 3, indicating a considerable expansion of the mesophases. Thus, during the heating scans, N phases were observed in the following order: 140.1 °C (1) > 104.4 °C (2) > 46.3 °C (3).UV-vis absorption
The UV-vis spectra for 1, 2, and 3 were measured in dichloromethane (DCM) and are shown in Fig. 2. For 1 and 3, the longest absorption edges are <400 nm, while that of 2 is ∼400 nm. These results indicate that 1–3 are suitable mesogenic units for optical applications, given that they exhibit low absorption in the visible wavelength region.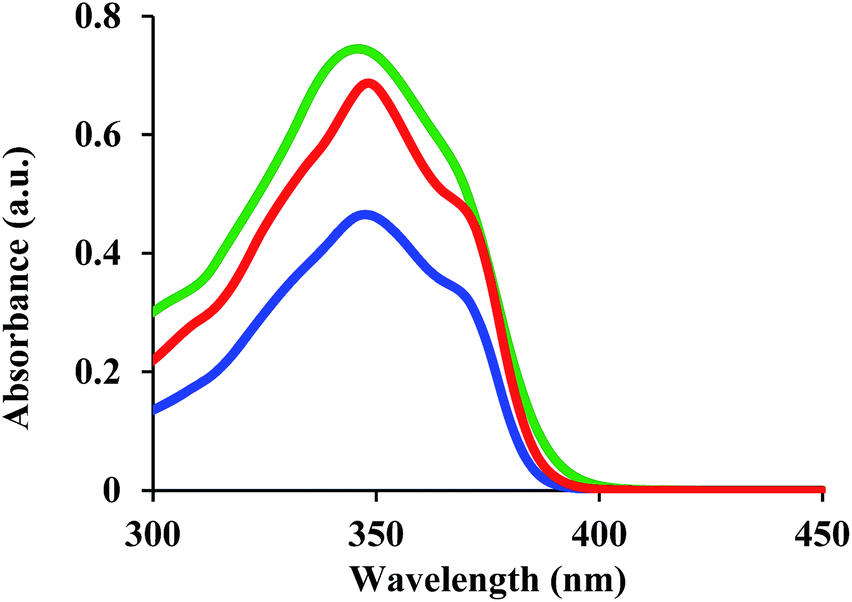 | ||
| Fig. 2 UV-vis spectra for 1 (red line), 2 (green line), and 3 (blue line) in DCM. | ||
Birefringence measurements
Subsequently, we evaluated the optical properties of 1–3. For that purpose, we measured ne, no, and calculated the corresponding Δn values (Δn = ne − no) from target material mediums, following a previously reported method.‡9,10 For 1, measurements were conducted up to T = 250 °C, as POM measurements showed signs of decomposition beyond this temperature, most likely on account of its high Ti. The representative transmittance spectra for 1 are shown in Fig. S7 and S8.† The temperature dependence of the obtained refractive index and Δn values at 550 nm are shown in Fig. 3, where TIN and T represent the transition temperatures from the isotropic (Iso) liquid to the N phase and the temperature measured upon cooling, respectively. Fig. 3(a) shows that 1–3 exhibited, on account of their highly anisotropic structures, their highly polarizable substituents, and their short alkyl chains, extremely high refractive index values (ne > 2.1). The corresponding value for 1 (ne > 2.3) is particularly interesting, as it is close to that of diamond. From a comparison of the temperature dependence of the ne and no values at TIN − T, it is apparent that the ne values of 1 and 2 are higher than that of 3, whereby the value of 1 is slightly higher than that of 2. In addition, the no values of 1 and 2 are also higher than that of 3, which explains the higher mean refractive index (〈n〉) of the former compounds with respect to the latter. The 〈n〉 values for 1 (1.84) and 2 (1.84) are comparable, and relatively high (Table 2). Fig. 3(b) illustrates the extremely high Δn values (>0.6) obtained for these compounds, which are a consequence of their high ne values. Notably, 1 reaches a maximum (Δn = 0.77) at the lowest measurement temperature (Table 2), which is, to the best of our knowledge, the highest Δn value so far obtained from a single component system, excluding extrapolated values from room-temperature nematic mixtures. This result demonstrates the success of the present design strategy, which is based on a combination of conjugation, asymmetry, and highly polarizable substituents.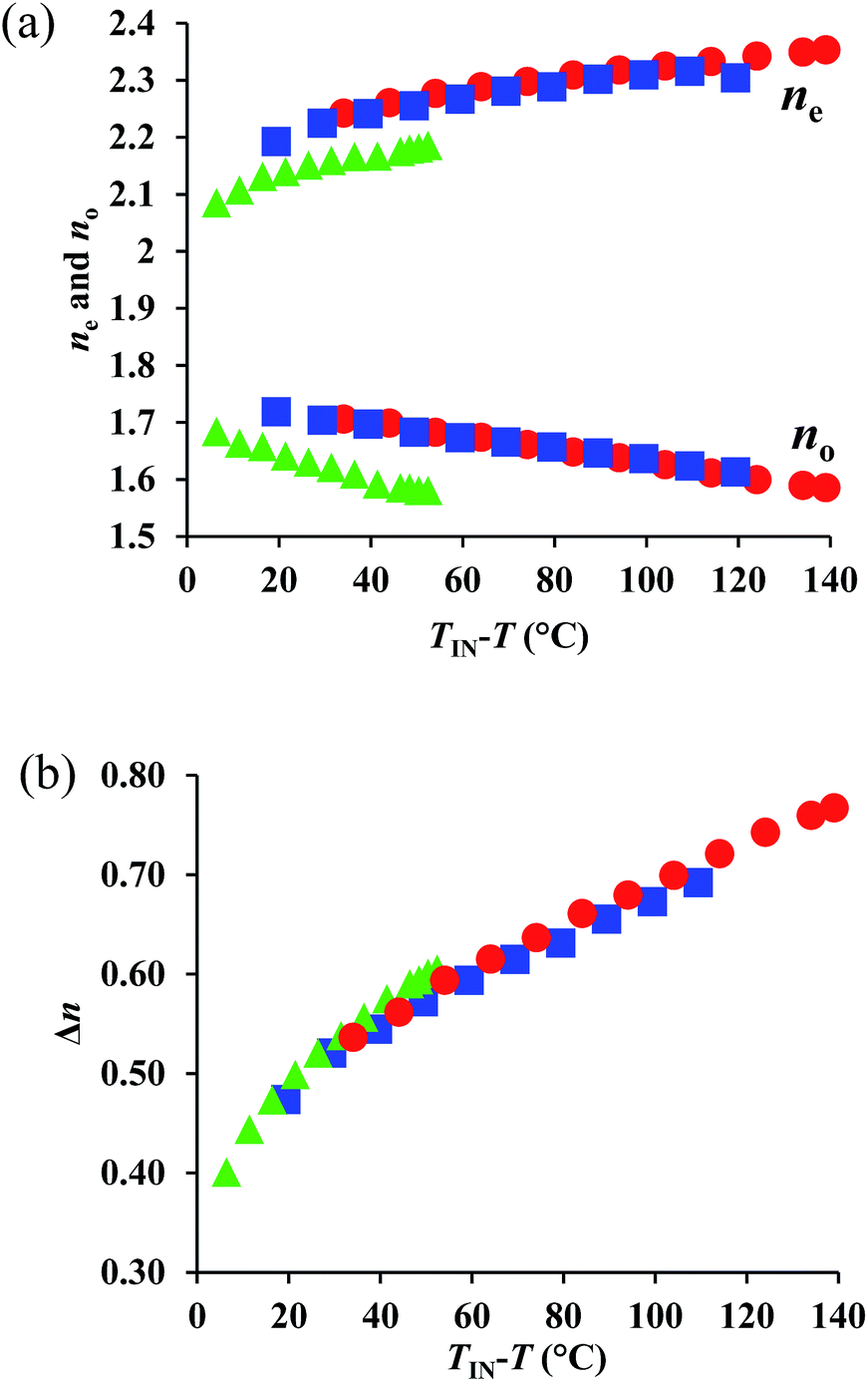 | ||
| Fig. 3 Temperature dependence of (a) ne and no, as well as (b) Δn values at 550 nm for 1 (red circles), 2 (red squares), and 3 (green triangles). | ||
In the high-temperature region (TIN − T < 30 °C), the temperature dependence of Δn for 1 and 2 is similar to that of 3. However, with increasing TIN − T, the Δn of 3 surpasses those of 1 and 2, despite the fact that the molecular polarizability anisotropy follows the order NCS > CN > SCH3. This result implies that the Δn of 3 increases with decreasing temperature at a higher rate than those of 1 and 2, which could be due to the enhanced S⋯S interactions of the two SCH3 (or SR) substituents in 3 (vide infra).
The wave-length dependences of the Δn values at each representative temperature are shown in Fig. S9–S11,† showing positive dispersion in analogy with those for typical rod-like molecules. We compared the ne, no, and Δn values for all compounds at 550 nm and 700 nm (Fig. 4). A comparison of Fig. 3 and 4 shows that the ne, no, and Δn values at 770 nm are lower than those at 550 nm, due to the positive dispersion of refractive indices for 1–3. Their temperature dependence at 700 nm exhibits a similar trend as that at 550 nm. For 1–3, the degree of wavelength dispersion, i.e., the Δn ratios at 550 nm and 700 nm (Δn (550 nm)/Δn (700 nm)), are ∼1.2. The comparable UV-vis absorptions (Fig. 2) suggest that the wave-length dependence of the refractive indices should not be affected substantially.
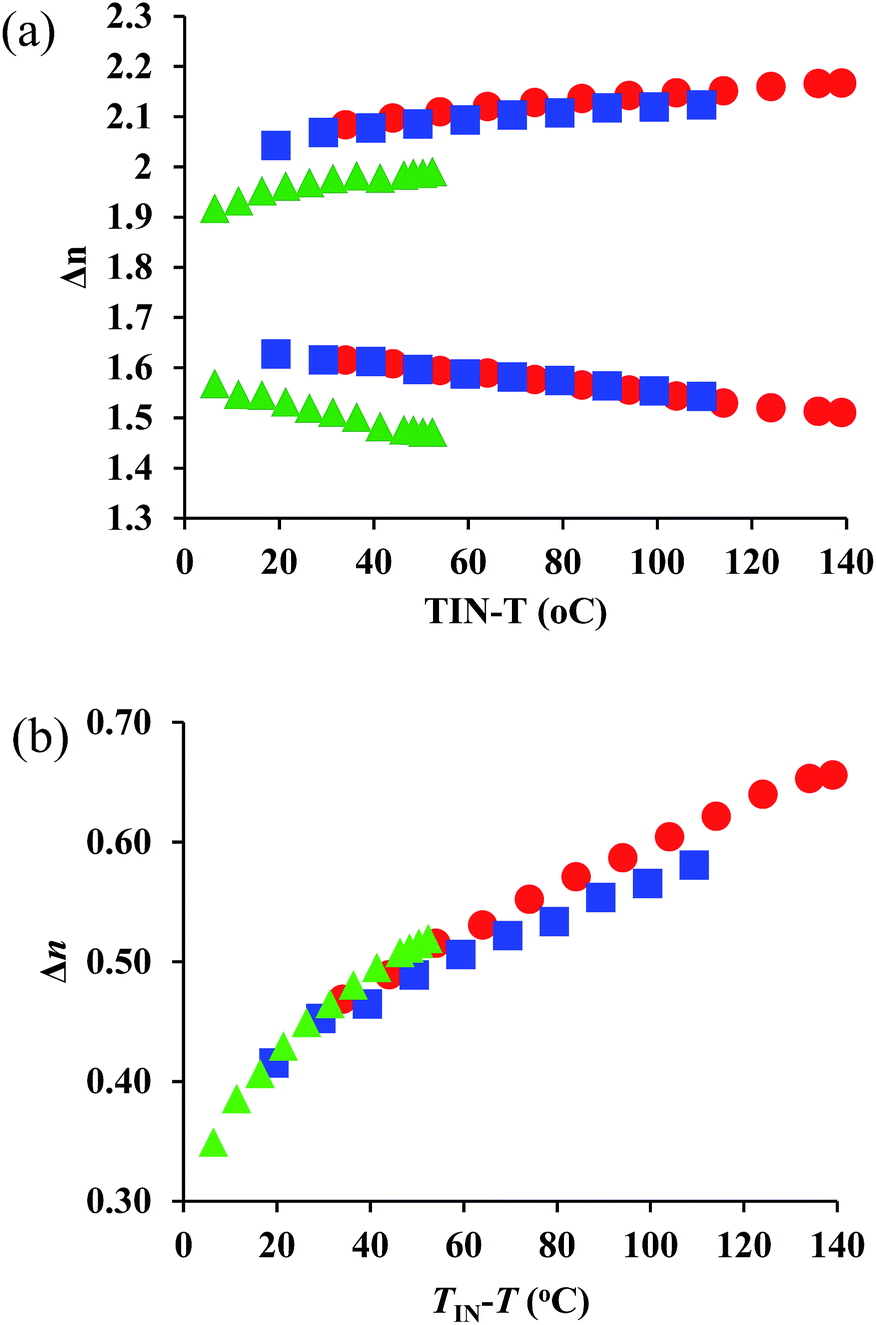 | ||
| Fig. 4 Temperature dependence of (a) ne and no, as well as (b) Δn values at 700 nm for 1 (red circles), 2 (red squares), and 3 (green triangles). | ||
WAXD measurements for the evaluation of the order parameters
Here, the temperature dependence of Δn is empirically described by the following equation: Δn = SΔno, wherein S = (1 − T/Ti)β; Δno refers to Δn for the perfectly oriented state, i.e., S = 1, while T and Ti represent measurement and clearing temperature, respectively; β is a constant characteristic for nematic materials.11 Accordingly, Δn is proportional to the order parameter S. Considering the aforementioned temperature dependence of Δn for each compound, we speculated that the determining factor may be the order parameters of 1–3. To examine the correlation between the order parameter and Δn, we estimated the temperature dependence of S by detailed XRD measurements using specimens aligned through an external magnetic field, which is described in detail elsewhere.12 The obtained 2D-XRD patterns are shown in Fig. S12–S14 (ESI).† The temperature dependence of S, using this wide-angle X-ray diffraction technique (WAXD), is shown in Fig. 5. The results demonstrate that these materials exhibit high-order parameters (S = ∼0.5) during the isotropic to nematic transition, and S > 0.7 near the crystallization temperature. These values are considerably higher than those for conventional rod-like LC molecules (S = ∼0.3–0.6).13 This is most likely due to the highly anisotropic structures of 1–3, which leads to higher degrees of anisotropic interactions. The wide mesophase of 1 (140 °C) induces a particularly high S value (0.75). Conversely, a comparison of the temperature dependence suggests that with decreasing temperature, the S value for 3 is much higher than those of 1 and 2, which means that this parameter is strongly influenced by temperature. Previously, we have reported that the optical properties of LC molecules with alkylsulfanyl groups exhibit a high temperature dependence as a consequence of the S⋯S interactions. Since the present results are consistent with this trend, and due to the S⋯S interactions between the two symmetric SCH3 substituents, the Δn values for 3 should be higher than those for 1 and 2 in the low temperature region (e.g. TIN − T = 30 °C). This result demonstrates that two symmetric SCH3 or SR substituents are able to induce higher levels of S⋯S interactions relative to an asymmetric substitution pattern with one SCH3 or SR substituent. This may lead to a higher Δn at similar TIN − T, even though 3 exhibited in the present case, on account of its narrower mesophase, a lower maximum Δn compared to 1 and 2.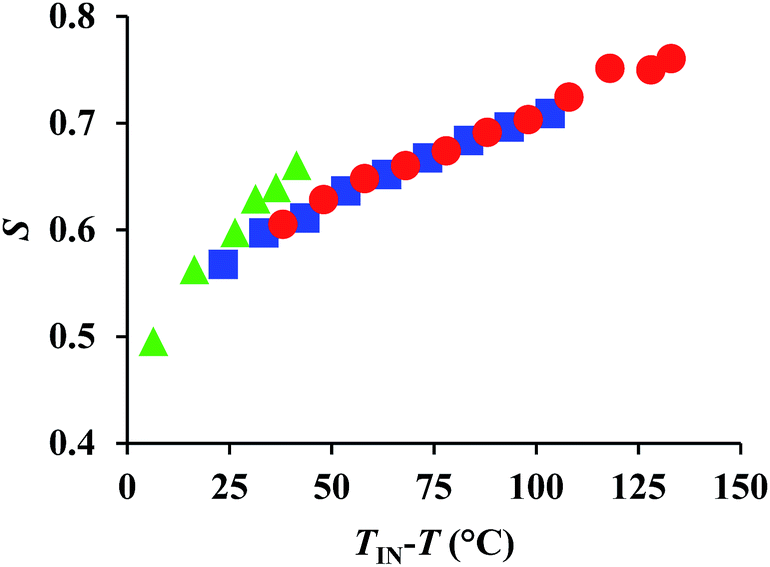 | ||
| Fig. 5 Temperature dependence of the estimated order parameter (S) obtained by WAXD measurement for 1 (red circles), 2 (blue squares), and 3 (green triangles). | ||
Conclusions
In summary, we presented the design and synthesis of three bistolane-based molecules with alkylsulfanyl groups (1–3). These materials exhibit well-defined enantiotropic nematic phases in the absence of any smectic phases. Owing to their asymmetric structure, the transition temperatures of 1 and 2 are significantly lower, and their nematic ranges are broader than those of symmetric 3. With respect to optical properties, it is noteworthy that 1–3 exhibit extremely high refractive indices and Δn values in their mesophases (e.g.1 and 2: ne > 2.3; 〈n〉 = ∼1.9). At the lowest measurement temperature, 1 showed an extremely high birefringence (Δn = 0.77). Moreover, 3 exhibited, due to the presence of two SR substituents, increased levels of S⋯S interactions relative to 1 and 2 with asymmetric substitution patterns including one SR group. This led to the highest Δn value for 3 close to TIN − T. Given their extremely high refractive index, these materials should find applications in various optical materials.Notes and references
- (a) J. W. Goodby, Liq. Cryst., 2011, 38, 1363 CrossRef CAS; (b) P. Kirsh and M. Bremer, Angew. Chem., Int. Ed., 2000, 39, 4216 CrossRef; (c) M. Mitov, Adv. Mater., 2012, 24, 6260 CrossRef CAS PubMed; (d) R. Dąbrowski, P. Kula and J. Herman, Crystals, 2013, 3, 443 CrossRef.
- (a) D. J. Broer, J. Lub and G. N. Mol, Nature, 1995, 378, 467 CrossRef CAS; (b) J. Hwang, M. H. Song, B. Park, S. Nishimura, T. Toyooka, J. W. Wu, Y. Takanishi, K. Ishikawa and H. Takezoe, Nat. Mater., 2005, 4, 383 CrossRef CAS PubMed; (c) M. Funahashi and N. Tamaoki, Mol. Cryst. Liq. Cryst., 2007, 475, 123 CrossRef CAS; (d) M. G. Chee, M. H. Song, D. Kim, H. Takezoe and I. J. Chung, Jpn. J. Appl. Phys., 2007, 46, L437 CrossRef CAS; (e) M. Itoh, M. Tokita, H. Hegi, T. Hayakawa, S. Kang and J. Watanabe, J. Mater. Chem., 2011, 21, 1697 RSC; (f) X. Chen, L. Wang, Y. Chen, C. Li, G. Hou, X. Liu, X. Zhang, W. He and H. Yang, Chem. Commun., 2014, 50, 691 RSCM. Uchimura, Y. Watanabe, F. Araoka, J. Watanabe, H. Takezoe and G. Konishi, Adv. Mater., 2010, 22, 4473 CrossRef CAS PubMed.
- (a) K. Okano, O. Tsutsumi, A. Shishido and T. Ikeda, J. Am. Chem. Soc., 2006, 128, 15368 CrossRef CAS PubMed; (b) K. Okano, A. Shishido, O. Tsutsumi, T. Shiono and T. Ikeda, J. Mater. Chem., 2005, 15, 3395 RSC; (c) K. Okano, A. Shishido and T. Ikeda, Adv. Mater., 2006, 18, 523 CrossRef CAS; (d) A. Shishido, Polym. J., 2010, 42, 525 CrossRef CAS; (e) N. Kawatsuki, H. Matsushita, T. Washio, J. Kozuki, M. Kondo, T. Sasaki and H. Ono, Macromolecules, 2014, 47, 324 CrossRef CAS.
- (a) H. R. Stapert, S. del Valle, E. J. K. Verstegen, B. M. I. van der Zande, J. Lub and S. Stallinga, Adv. Funct. Mater., 2003, 13, 732 CrossRef CAS; (b) P. Valley, D. L. Mathine, M. R. Dodge, J. Schwiegerling, G. Peyman and N. Peyghambarian, Opt. Lett., 2010, 35, 336 CrossRef PubMed; (c) P. Valley, M. R. Dodge, J. Schwiegerling, G. Peyman and N. Peyghambarian, Opt. Lett., 2010, 35, 2582 CrossRef PubMed.
- (a) D. Węgłowska, P. Kula and J. Herman, RSC Adv., 2016, 6, 403 RSC; (b) S. Gauza, C.-H. Wen, S.-T. Wu, N. Janarthanan and C.-S. Hsu, Jpn. J. Appl. Phys., 2004, 43, 7634 CrossRef CAS; (c) C. Sekine, M. Ishitobi, K. Iwakura, M. Minai and K. Fujisawa, Liq. Cryst., 2002, 29, 355 CrossRef CAS; (d) X.-L. Guan, L.-Y. Zhang, Z.-L. Zhang, Z. Shen, X.-F. Chen, X.-H. Fan and Q.-F. Zhou, Tetrahedron, 2009, 65, 3728 CrossRef CAS; (e) Z. Zhang, L. Zhang, X. Guan, Z. Shen, X. Chen, G. Xing, X. Fan and Q. Zhou, Liq. Cryst., 2010, 37, 69 CrossRef CAS.
- (a) Y. Arakawa, S. Nakajima, R. Ishige, M. Uchimura, S. Kang, G. Konishi and J. Watanabe, J. Mater. Chem., 2012, 22, 8394 RSC; (b) S.-T. Wu, U. Finkenzeller and V. Reiffenrath, J. Appl. Phys., 1989, 65, 4372 CrossRef CAS; (c) S.-T. Wu, C.-S. Hsu and K.-F. Shyu, Appl. Phys. Lett., 1999, 74, 344 CrossRef CAS; (d) A. Spadło, R. Dąbrowski, M. Filipowicz, Z. Stolarz, J. Przedmojski, S. Gauza, C. Y. H. Fan and S.-T. Wu, Liq. Cryst., 2003, 30, 191 CrossRef; (e) M. Hird, A. J. Seed, K. J. Toyne, J. W. Goodby, G. W. Gray and D. G. McDonnell, J. Mater. Chem., 1993, 3, 851 RSC; (f) Y.-M. Zhang, D. Wang, Z.-C. Miao, S.-K. Jin and H. Yang, Liq. Cryst., 2012, 39, 1330 CrossRef CAS; (g) C. Sekine, N. Konya, M. Minai and K. Fujisawa, Liq. Cryst., 2001, 28, 1361 CrossRef CAS; (h) O. Catanescu and L.-C. Chien, Liq. Cryst., 2006, 33, 115 CrossRef CAS; (i) P. Kula, J. Herman, S. Pluczyk, P. Harmata, G. Mangelinckx and J. Beeckman, Liq. Cryst., 2014, 41, 503 CrossRef CAS; (j) D. V. Sai, P. Sathyanarayana, V. S. S. Sastry, J. Herman, P. Kula, R. Dabrowski and S. Dhara, Liq. Cryst., 2014, 41, 591 CrossRef; (k) Y. Arakawa, S. Kang, S. Nakajima, K. Sakajiri, Y. Cho, S. Kawauchi, J. Watanabe and G. Konishi, J. Mater. Chem. C, 2013, 1, 8094 RSC; (l) Y. Arakawa, S. Nakajima, S. Kang, M. Shigeta, G. Konishi and J. Watanabe, J. Mater. Chem., 2012, 22, 13908 RSC; (m) Y. Arakawa, S. Nakajima, S. Kang, M. Shigeta, G. Konishi and J. Watanabe, Liq. Cryst., 2012, 39, 1063 CrossRef CAS; (n) Y. Arakawa, S. Kang, J. Watanabe and G. Konishi, Chem. Lett., 2014, 43, 1858 CrossRef; (o) Y. Arakawa, H. Kuwahara, K. Sakajiri, S. Kang, M. Tokita and G. Konishi, Liq. Cryst., 2015, 42, 1419 CrossRef CAS; (p) Y. Arakawa, S. Kang, J. Watanabe and G. Konishi, Phase Transitions, 2015, 88, 1188 CrossRef; (q) E.-W. Lee, M. Hattori, F. Uehara, M. Tokita, S. Kawauchi, J. Watanabe and S. Kang, J. Mater. Chem. C, 2015, 3, 2266 RSC; (r) Y.-Z. Zhao, D. Wang, Z.-M. He, G. Chen, L.-Y. Zhang, H.-Q. Zhang and H. Yang, Chin. Chem. Lett., 2015, 26, 785 CrossRef CAS; (s) Y. Arakawa, S. Nakajima, S. Kang, G. Konishi and J. Watanabe, J. Mater. Chem., 2012, 22, 14346 RSC.
- Y. Arakawa, S. Kang, H. Tsuji, J. Watanabe and G. Konishi, RSC Adv., 2016, 6, 16568 RSC.
- (a) A. J. Seed, K. J. Toyne, J. W. Goodby and D. G. McDonnell, J. Mater. Chem., 1995, 5, 1 RSC; (b) A. J. Seed, K. J. Toyne and J. W. Goodby, J. Mater. Chem., 1995, 5, 2201 RSC; (c) G. J. Cross, A. J. Seed, K. J. Toyne, J. W. Goodby, M. Hird and M. C. Artal, J. Mater. Chem., 2000, 10, 1555 RSC; (d) A. J. Seed, K. J. Toyne, M. Hird and J. W. Goodby, Liq. Cryst., 2012, 39, 403 CrossRef CAS; (e) A. J. Seed, K. Pantalone, U. M. Sharma and A. M. Grubb, Liq. Cryst., 2009, 36, 329 CrossRef CAS; (f) Y. Arakawa, S. Kang, J. Watanabe and G. Konishi, RSC Adv., 2015, 5, 8056 RSC; (g) D. Węgłowska, P. Kula and J. Herman, RSC Adv., 2016, 6, 403 RSC.
- (a) R. Chang, Mol. Cryst. Liq. Cryst., 1974, 28, 1 CrossRef CAS; (b) E. Hecht, Optics, Addison-Wesley, Boston, 4th edn, 2002, pp. 416–425 Search PubMed.
- (a) S. Kang, S. Nakajima, Y. Arakawa, G. Konishi and J. Watanabe, J. Mater. Chem. C, 2013, 1, 4222 RSC; (b) Y. Arakawa, S. Kang, S. Nakajima, K. Sakajiri, S. Kawauchi, J. Watanabe and G. Konishi, Liq. Cryst., 2014, 41, 642 CrossRef CAS.
- I. Haller, Prog. Solid State Chem., 1975, 10, 103 CrossRef.
- (a) Handbook of Liquid Crystals, WILEY-VCH Verlag GmbH & Co. KGaA, 2014, vol. 1, pp. 317–318 Search PubMed; (b) S. Kang, S. Nakajima, Y. Arakawa, M. Tokita, J. Watanabe and G. Konishi, Polym. Chem., 2014, 5, 2253 RSC; (c) K. Fu, M. Sone, M. Tokita and J. Watanabe, Polym. J., 2006, 38, 442 CrossRef CAS.
- (a) I. Dierking, Textures of Liquid Crystals, WILEY-VCH Verlag GmbH & Co. KGaA, 2003 CrossRef; (b) D. H. Chen and G. R. Luckhurst, Trans. Faraday Soc., 1969, 65, 656 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization, DSC chats, POM photos, refractive index, and WAXD measurements. See DOI: 10.1039/c6ra14093a |
| ‡ As we could not observe clear transmittance spectra, ne values for 3 were estimated from ne = Δn − no. In this case, the temperature dependence of Δn was evaluated according to a previously described method; see: ref. 6a. |
| This journal is © The Royal Society of Chemistry 2016 |