
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From mononuclear to linear one-dimensional coordination species of copper(II)–chloranilate: design and characterization†
Marijana
Jurić
,
Krešimir
Molčanov
*,
Dijana
Žilić
and
Biserka
Kojić-Prodić
Ruđer Bošković Institute, Bijenička 54, HR-10000 Zagreb, Croatia. E-mail: kmolcano@irb.hr
First published on 22nd June 2016
Abstract
A series of six novel mononuclear, binuclear and linear one-dimensional (1D) compounds of copper(II) with chloranilic acid (3,6-dichloro-2,5-dihydroxybenzoquinone, H2CA) is prepared and a design strategy for the preparation of such complexes is discussed. Four described compounds are linear 1D coordination polymers [Cu(CA)]n, whereas another two involve a binuclear and a mononuclear, Cu2(CA)3 and Cu(CA)2, core unit. A linear polymer incorporating bulky aromatic imidazole has been synthesized as a result of investigation of the influence of pH on the reaction mixture. Two coordination modes of the chloranilate dianion are observed. The bridging (bis)bidentate mode generates linear 1D polymeric species. Among these one reveals square-pyramidal coordination of Cu2+, whereas the three polymers contain Cu2+ in an octahedral arrangement. However, the combination of both, terminal bidentate (ortho-quinone) and bridging (bis)bidentate modes of coordination produces a binuclear complex anion, which comprises a square-pyramidal coordination of Cu2+ complex anions forming a supramolecular honeycomb-like network encapsulating 4,4′-bipyridine cations. When the chloranilate dianion coordinates the Cu2+ atom only in a terminal bidentate mode, a mononuclear complex with an octahedral environment of the metal centre is formed. The presence of the bulky ancillary ligand imidazole produces an unprecedented packing involving chiral (racemic) and achiral (meso-compound) coordination polymers in the same crystal. Electron spin resonance spectroscopy of polycrystalline samples determined g-tensor parameters of copper(II) ions in different coordination geometries and revealed weak exchange interactions (|J| < 1 cm−1) in linear metal-complex polymers and dimeric species.
Introduction
Coordination polymers, also known as metal–organic coordination networks (MOCNs) or metal–organic frameworks (MOFs), are metal–ligand compounds having infinite one- (1D), two- (2D) or three-dimensional (3D) networks through more or less covalent metal–ligand bonding. The ligand should be a bridging organic group and the metal atoms must only be bridged by this organic ligand at least in one dimension.1,2 Due to the diversity of metal species and ligands, coordination geometry, guests inside the pores, and supramolecular arrangements, a huge number of coordination polymers with different structures and pores have been synthesized and characterized; different dimensionality and topology of the metal complexes result in a variety of magnetic and/or electrical properties.2 The magnetic properties can be designed and fine-tuned by choice of the bridging ligand; small ligand strongly affects exchange interaction and spin coupling, whereas some ligands with conjugated π-electron systems are able to mediate exchange interactions at long distances. The careful selections of the organic ligands used for a fine tuning of the physical properties lead to various applications, such as catalysis, electrical conductivity, luminescence, magnetism, non-linear optics or zeolite behaviour. Generally, the use of nitrogen- and oxygen-donor ligands prevails in the construction of coordination polymers.3–5 Selection of the metal cation(s) offers further options for design of magnetic properties. Also, a good combination of metal(s) and ligand(s) may lead to a coordination polymer which undergoes a reversible magnetic transition under appropriate external stimulus (temperature, pressure, irradiation by a specific wavelength).6–9 Besides magnetic properties, coordination polymers may possess electrical properties: conductivity, semiconductivity, ferroelectricity, etc. Electronic conductivity is possible along a polymeric chain comprising ligands with conjugated π-systems. Design of such systems and fine-tuning of the interactions defining the properties, is a topic of crystal engineering.2,10,11Substituted 2,5-dihydroxybenzoquinonates (2,5-DHQs), such as 3,6-dichloro-2,5-dihydroxybenzoquinone (chloranilic acid, H2CA) are among the most versatile ligands for construction of the coordination polymers with different topologies.11–20 They are prone to form a variety of coordination modes19–24 and to mediate electronic effects between paramagnetic metal ions via their four oxygen donors and π-electron systems, respectively.25 Usually 2,5-dihydroxyquinones are bridging (bis)bidentate ligands, but sometimes may also act as bidentate + bridging monodentate ligands,26 with the possibility to generate polymeric complexes. Mononuclear complexes with chloranilic acid acting as a terminal bidentate ligand are known as well.26,27
Actually, 2,5-DHQs can be regarded as “extended” (i.e. sterically bulkier) version of the oxalate.26 Conjugated bonds of the quinoid ring and presence of two acidic hydroxy-groups make the electronic structure of 2,5-DHQs26,27 uniquely malleable depending on ionisation, molecular environment, and degree of electron delocalisation characteristics; it can exist as an ortho- or para-quinoid structure, or a dianion-like structure with two separated delocalised systems (Scheme 1). Quinones are also weak oxidants, and their proton-accepting properties can be enhanced by electronegative substituents, thus enabling the charge transfer between a metal centre and the quinoid ring; the quinone is then reduced into a semiquinone radical.28,29 Their potential in design of magnetic, charge-transfer and spin-crossover materials has been little studied so far.
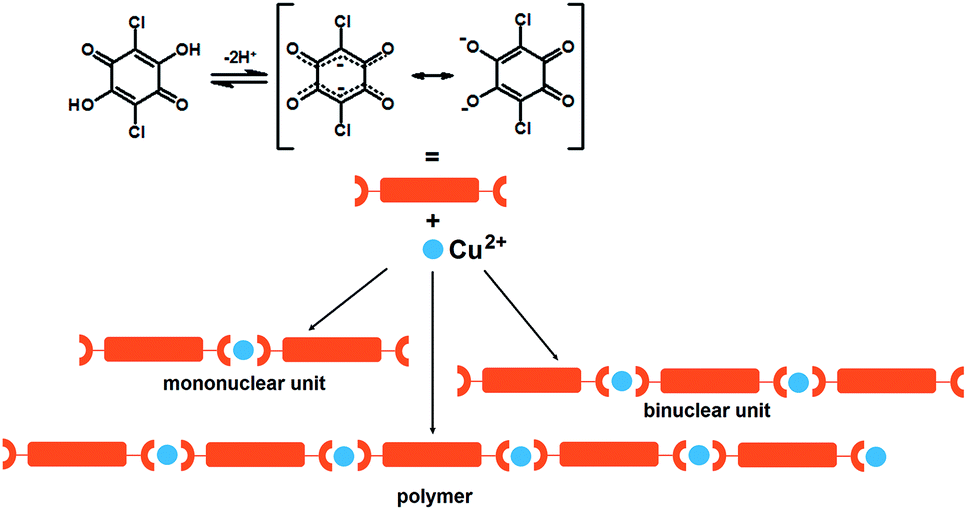 | ||
| Scheme 1 Dissociation of chloranilic acid and schematic representation of its binding to copper(II) ions investigated in this work. | ||
The rational design and optimisation of synthesis of the coordination polymers based on chloranilic acid and first-row transition metals is the goal of our research. The use of bridging mode of CA ligand should be in favour of formation of linear Cu(II) polymeric chains due to its ability to mediate electronic effects between paramagnetic metal ions, even at large Cu⋯Cu separation distance (exceeding 7 Å). The size of the metal cation is an important parameter: larger cations (for example: Cr3+, Mn2+, Fe3+) can better accommodate large multidentate ligands than smaller ones (Cu2+, Zn2+). Therefore, Cu2+ complexes with CA and bidentate N-donor ligands are almost exclusively mononuclear,26,27 while larger cations can accommodate zig–zag 1D polymers.22,30 Linear 1D-polymers of Cu2+ are obtained if additional bidentate ligands were replaced by solvent molecules such as water31 and methanol13 acting as monodentate ancillary ligands. An appropriate bridging (bis)monodentate ligand, such as pyrazine, can link the chains into a 2D network.13
Results and discussion
We designed and prepared a series of six novel compounds: mononuclear (Him)2[Cu(CA)2(H2O)2] (1; im = imidazole, C3H4N2), binuclear (4,4′-H2bpy)[Cu2(CA)3(H2O)2]·2H2O (2; 4,4′-bpy = 4,4′-bipyridine, C10H8N2), and 1D coordination polymers [Cu(CA)(CH3CN)]n (3), [Cu(CA)(H2O)2]n (4), [Cu(CA)(EtOH)2]n (5) and [Cu(CA)(im)2]n (6) (Scheme 1). They are studied using the X-ray structural analysis, IR and ESR spectroscopies, performed to obtain spin-Hamiltonian parameters of copper(II) ions as well as the values of exchange interactions in the compounds. The dark crystals of the titled compounds were obtained by slow liquid diffusion. This layering technique was applied for the preparation of the crystals of the quality needed for X-ray analysis.32Molecular structures of compounds 1–6
The compound, 1, is a salt of a mononuclear complex anion [Cu(CA)2(H2O)2]2− (Fig. 1) with imidazole as cations (Him+); its asymmetric unit comprises a half of the complex anion with molecular symmetry Ci (Fig. 1) and a single imidazolium cation (Fig. S1†). The central copper atom is located at an inversion centre (1/2, 0, 1/2) and its coordination is a distorted octahedron (Table S1†).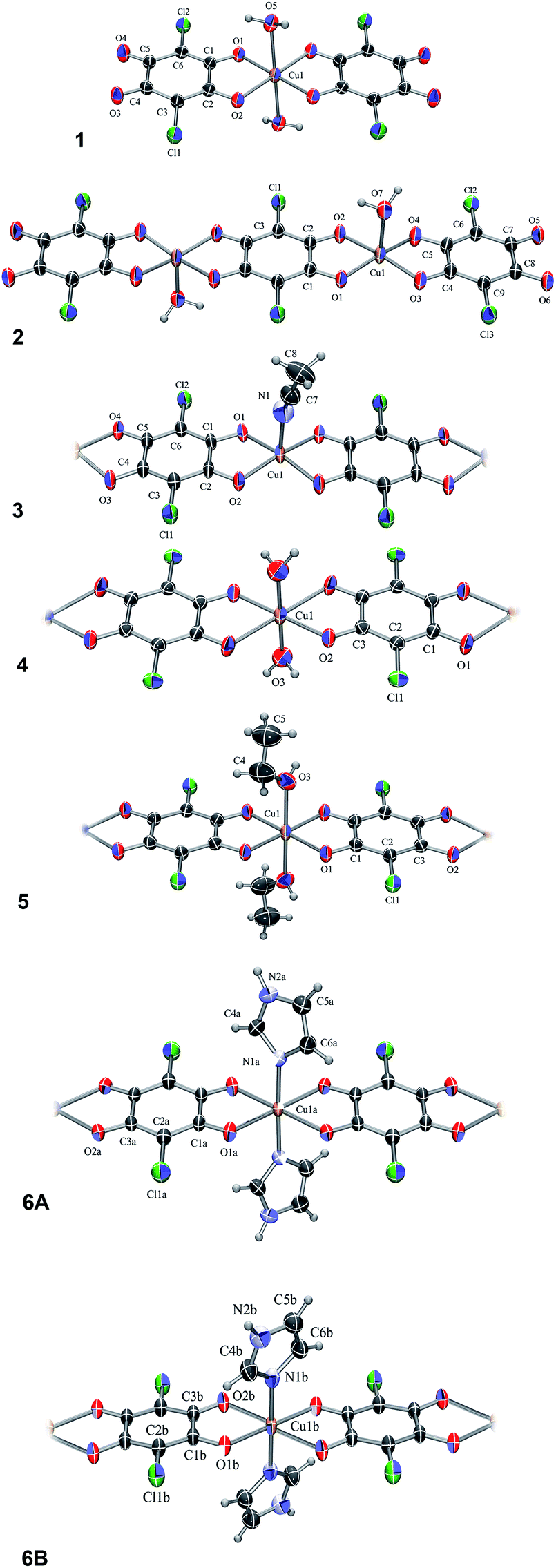 | ||
| Fig. 1 ORTEP-3 (ref. 33) drawings of the Cu–chloranilate complex units: [Cu(CA)2(H2O)2]2− (1), [Cu2(CA)3(H2O)2]2− (2), [Cu(CA)(CH3CN)]n, (3), [Cu(CA)(H2O)2]n (4), [Cu(CA)(EtOH)2]n (5), a C2-symmetric chain A and achiral centrosymmetric chain B of [Cu(CA)(im)2]n (6A and 6B, respectively). Displacement ellipsoids are drawn at the probability of 50% and hydrogen atoms are shown as spheres of arbitrary radii. | ||
The compound 2 is the complex salt of a dinuclear anion with 4,4′-bpy cation and crystal water molecules (4,4′-H2bpy)[Cu2(CA)3(H2O)2]·2H2O. Its asymmetric unit is a half of the dianion (Fig. 1) and a half of the cation (Fig. S2†), both having a Ci symmetry. The coordination of copper atom is a distorted square-pyramid with a water molecule at the apical position (Table S1†).
In the polymer 3, [Cu(CA)(CH3CN)]n (Fig. 1), copper coordination is also a distorted square-pyramid (Table S1†), where its apical position is occupied by an acetonitrile molecule. This is the first [Cu(CA)]n coordination polymer with pentacoordinated copper(II) ion; so far pentacoordination of Cu2+ was observed in mononuclear species26,27 and three dinuclear complexes containing bridging CA ligand.34–36
The analogous compounds 4, 5 and 6 consist of linear 1D chains with chemical compositions of [Cu(CA)(H2O)2]n, [Cu(CA)(EtOH)2]n and [Cu(CA)(im)2]n, respectively (Fig. 1). The Cu atom and chloranilate ring have crystallographic symmetry Ci in 4 and C2 in 5. In all these complexes, copper coordination is a distorted octahedron (Table S1†): the metal is coordinated by two chloranilates in the equatorial position (in a trans arrangement) and two water/ethanol/imidazole molecules occupying two apical positions. Previously prepared and investigated compound {[Cu(CA)(H2O)2]·H2O}n (ref. 31) contains the same polymeric unit as our compound 4, however, it is a monohydrate, while we report the structure without a crystal water.
In 6 exist two symmetry-independent chains (designated as A and B; Fig. 1), both parallel to [010] direction. The chain A is C2-symmetric, with twofold axis passing through Cu atoms and midpoints of chloranilate rings. It is therefore chiral, with both R and S enantiomers (Scheme 2) present due to the centrosymmetric space group. The chain B is centrosymmetric, with Cu atoms and chloranilate rings having a crystallographic symmetry Ci. It can be regarded as a meso-compound, and differs from its diastereomer A in orientation of imidazole ligands, as seen in Fig. 2 and Scheme 2. This is a rare example of co-crystallization of a chiral and a meso-molecule.37
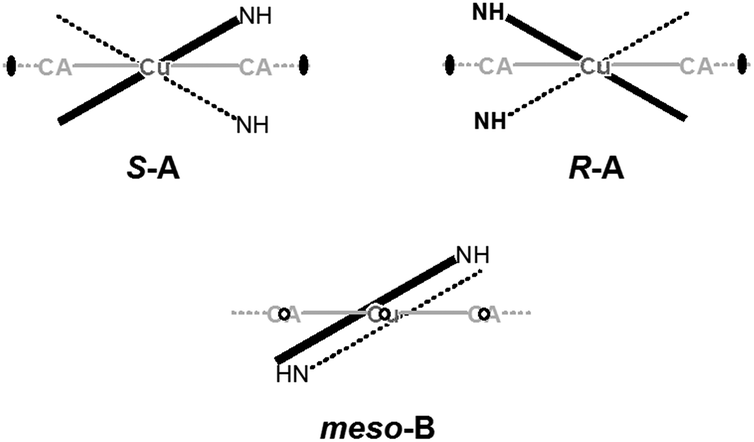 | ||
| Scheme 2 Schematic representation of symmetry in chiral chains (imidazole moieties are related by a twofold axis) and meso-chains (imidazole moieties are related by inversion). Direction of the [Cu(CA)]n ribbon of 6 has been indicated in gray. | ||
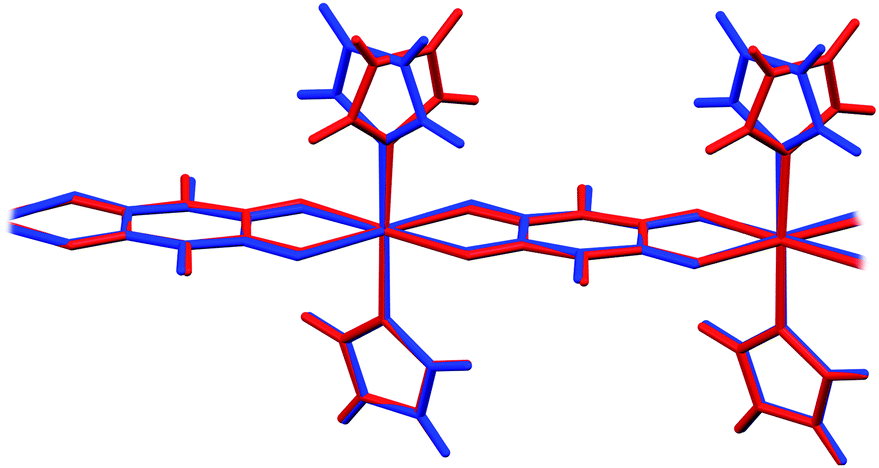 | ||
| Fig. 2 Overlay of two symmetry-independent chains in [Cu(CA)(im)2]n (6). Chiral C2-symmetric chain A is red, while the achiral meso-chain B is blue. | ||
In four prepared polymers chloranilate dianions act as bridging (bis)bidentate ligands, and have dianion-like geometry with two delocalized systems separated by two single C–C bonds (Scheme 1 and Table 1). In compounds 1 and 2, two chloranilates act as terminal bidentate ligands and have an ortho-quinone-like geometry, while the central one in 2 acts as a bridging (bis)bidentate ligand and has dianion-like characteristics (Table 1).26,27 So far, only a few compounds are known that comprise two different coordination modes of chloranilic acid,15,17,24,38–40 which could be easily distinguished by IR spectra.
| 1 | 2-Terminal | 2-Bridging | ||||||
|---|---|---|---|---|---|---|---|---|
| Single | C1–O1 | 1.283(3) | Single | C4–O3 | 1.286(3) | Delocalised | C1–O1 | 1.261(3) |
| Single | C2–O2 | 1.278(3) | Single | C5–O5 | 1.278(3) | Delocalised | C3–O2 | 1.261(3) |
| Double | C4–O3 | 1.228(3) | Double | C7–O5 | 1.238(3) | Delocalised | C1–C2 | 1.391(3) |
| Double | C5–O4 | 1.224(3) | Double | C8–O6 | 1.229(3) | Delocalised | C2–C3 | 1.398(3) |
| Single | C1–C2 | 1.524(4) | Single | C4–C5 | 1.506(3) | Single | C1–C3i | 1.552(3) |
| Double | C2–C3 | 1.370(4) | Double | C4–C9 | 1.375(3) | |||
| Single | C3–C4 | 1.410(4) | Double | C5–C6 | 1.379(3) | |||
| Single | C4–C5 | 1.554(4) | Single | C6–C7 | 1.419(3) | |||
| Single | C5–C6 | 1.424(4) | Single | C7–C8 | 1.554(3) | |||
| Double | C6–C1 | 1.367(4) | Single | C8–C9 | 1.424(3) | |||
| 3 | 4 | 5 | ||||
|---|---|---|---|---|---|---|
| Delocalised | C1–O1 | 1.269(4) | Delocalised | C1–O1 | 1.252(9) | 1.254(4) |
| Delocalised | C2–O2 | 1.257(4) | Delocalised | C3–O2 | 1.273(8) | 1.259(4) |
| Delocalised | C4–O3 | 1.264(4) | Delocalised | C1–C2 | 1.404(10) | 1.383(4) |
| Delocalised | C5–O4 | 1.265(4) | Delocalised | C2–C3 | 1.372(9) | 1.382(5) |
| Single | C1–C2 | 1.527(5) | Single | C1–C3i | 1.533(9) | 1.547(4) |
| Delocalised | C2–C3 | 1.397(4) | ||||
| Delocalised | C3–C4 | 1.382(4) | ||||
| Single | C4–C5 | 1.524(5) | ||||
| Delocalised | C5–C6 | 1.396(4) | ||||
| Delocalised | C6–C1 | 1.376(4) | ||||
| 6a | 6b | |||
|---|---|---|---|---|
| Delocalised | C1–O1 | 1.248(2) | C1–O1 | 1.257(2) |
| Delocalised | C3–O2 | 1.256(2) | C3–O2 | 1.244(2) |
| Delocalised | C1–C2 | 1.401(2) | C1–C2 | 1.392(2) |
| Delocalised | C2–C3 | 1.392(2) | C2–C3 | 1.407(2) |
| Single | C3–C3ii | 1.539(3) | C1–C3i | 1.536(3) |
Generally, the positions of the symmetric and asymmetric carboxylate stretching vibrations in the IR spectrum are indicative of the binding mode of the ligand. Selected absorption bands ascribed to the vibrations of the bidentate26,27 and bis(bidentate)11,41–43 chloranilate anion for compounds 1–6 are given in Table 2. In terminal bidentate ligand, single and double C–O bonds can be distinguished,26,27 with their respective stretching vibrations close to the neutral chloranilic acid.44,45 In the case of bridging (bis)bidentate ligand, whose structure is similar to that of the free dianion, only one type of the stretching vibration of the C–O bond is present in the spectrum, consistent with bond length about 1.26 Å (in between single and double bond, Table 1). The spectra of 1 and 2 demonstrate some differences (Table 2); in compound 1 chloranilate anion has bidentate coordination mode so stretching vibrations of ν(C–O) and ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) could be observed, while in compound 2, having bidentate and bridging bis(bidentate) chloranilate anions, ν(C–O), ν(CO) and ν(C⋯O). In compounds 3–6 chloranilate anion is bridging and only [ν(C⋯O)] is found (Table 2). In the spectra of compounds 1, 2 and 6 other absorption bands of significant intensities correspond to different vibrations of the imidazole and 4,4′-bipyridine molecules.46
O) could be observed, while in compound 2, having bidentate and bridging bis(bidentate) chloranilate anions, ν(C–O), ν(CO) and ν(C⋯O). In compounds 3–6 chloranilate anion is bridging and only [ν(C⋯O)] is found (Table 2). In the spectra of compounds 1, 2 and 6 other absorption bands of significant intensities correspond to different vibrations of the imidazole and 4,4′-bipyridine molecules.46
| Compound | ν(C⋯O) |
ν(CO) |
ν(C–O) | ν(C–Cl) | C–Cl wagging |
|---|---|---|---|---|---|
| 1 | — | 1640, 1597 | 1340 | 851 | 569 |
| 2 | 1508, 1366 | 1637, 1609 | 1337 | 866, 847 | 573 |
| 3 | 1490, 1370 | — | — | 866 | 579 |
| 4 | 1507, 1380 | — | — | 856 | 579 |
| 5 | 1486, 1368 | — | — | 866 | 580 |
| 6 | 1512, 1370 | — | — | 851 | 576 |
Crystal packing of compounds 1–6
In compound 1 complex anions [Cu(CA)2(H2O)2]2− form hydrogen bonded layers parallel to (110), which are linked through imidazolium cations into a 3D network (Fig. 3a, Table 3). There are infinite π-stacks parallel to [001]: stacks of chloranilate moieties stabilise the anionic layers, whereas the imidazolium cations form their own stacks (Fig. 3b, Table 4). This structure came as a surprise, since imidazole was intended as a ligand (due to its two nitrogens, it may be able to link the [CuCA]n chains, similar to pyrazine13), rather than a counter-ion; however, its coordination to Cu2+ required a more basic solution.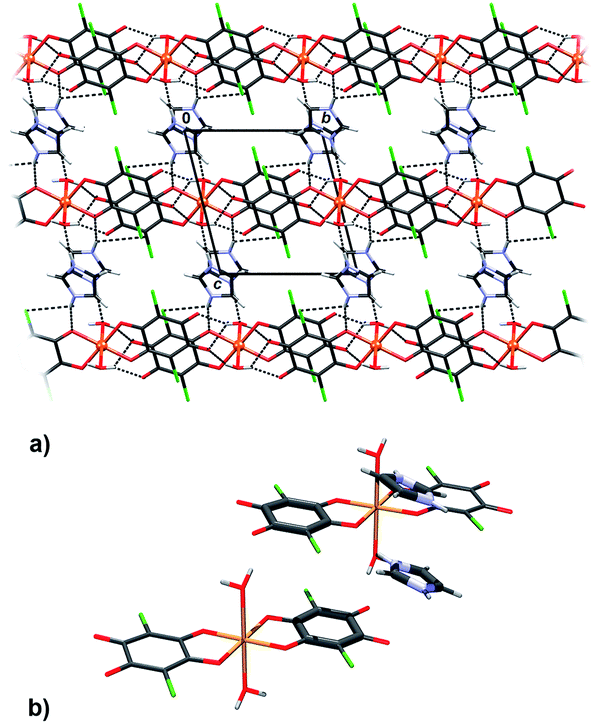 | ||
| Fig. 3 Crystal packing of 1 showing (a) layers of hydrogen-bonded [Cu(CA)2(H2O)2]2− anions linked through imidazolium cations into a 3D network and (b) π-stacking between two complex anions and two imidazolium cations. | ||
| D–H⋯A | D–H/Å | H⋯A/Å | D⋯A/Å | D–H⋯A/° | Symm. op. on A |
|---|---|---|---|---|---|
| 1 | |||||
| N1–H1⋯O1 | 0.86 | 2.04 | 2.867(3) | 161 | x, y, z |
| N2–H2⋯O5 | 0.86 | 1.96 | 2.810(3) | 172 | 1 − x, −y, 2 − z |
| O5–H5A⋯O4 | 0.92(4) | 1.90(4) | 2.794(3) | 162(4) | x, −1 + y, z |
| O5–H5B⋯O3 | 0.92(3) | 1.86(3) | 2.692(3) | 149(3) | −x, 1 − y, 1 − z |
| O5–H5B⋯O4 | 0.92(3) | 2.58(3) | 3.325(3) | 139(3) | −x, 1 − y, 1 − z |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| 2 | |||||
| N1–H1⋯O8 | 0.86 | 1.95 | 2.724(4) | 148 | 1 − x, 1 − y, 2 − z |
| O7–H7A⋯O5 | 0.94(3) | 1.89(3) | 2.811(3) | 165(3) | −x, 1 − y, 1 − z |
| O7–H7B⋯O5 | 0.92(5) | 1.90(3) | 2.784(3) | 161(4) | 1 + x, y, z |
| O8–H8A⋯O3 | 0.94(4) | 2.34(4) | 3.228(3) | 157(4) | x, y, z |
| O8–H8A⋯O7 | 0.94(4) | 2.42(4) | 2.948(3) | 116(4) | x, y, z |
| O8–H8B⋯Cl3 | 0.92(4) | 2.79(5) | 3.518(3) | 136(4) | −x, 1 − y, 2 − z |
| O8–H8B⋯O6 | 0.92(4) | 2.21(3) | 2.965(3) | 138(4) | −x, 1 − y, 2 − z |
|
|||||
| 3 | |||||
| C8–H8A⋯O3 | 0.94(4) | 2.73(6) | 3.367(7) | 130(7) | 1 − x, −y, 1 − z |
| C8–H8A⋯O4 | 0.94(4) | 2.63(5) | 3.119(6) | 113(3) | −1 + x, y, z |
|
|||||
| 4 | |||||
| O3–H3A⋯Cl1 | 0.95(10) | 2.61(10) | 3.492(7) | 155(7) | −x, −1/2 + y, 3/2 − z |
| O3–H3B⋯O1 | 0.95(9) | 2.55(10) | 3.107(9) | 118(6) | x, −1 + y, z |
|
|||||
| 5 | |||||
| O3–H3⋯O2 | 0.81(4) | 2.15(4) | 2.940(4) | 167(7) | x, −y, 1/2 + z |
| O3–H3⋯Cl1 | 0.81(4) | 2.91(7) | 3.395(4) | 121(5) | x, −y, 1/2 + z |
|
|||||
| 6 | |||||
| N2A–H2A⋯O2B | 0.86 | 2.10 | 2.833(2) | 143 | 1/2 + x, 3/2 − y, 1/2 + z |
| N2B–H2B⋯O2A | 0.86 | 2.16 | 2.879(3) | 141 | x, 1 + y, z |
| N2B–H2B⋯Cl1A | 0.86 | 2.90 | 3.503(3) | 128 | 1 − x, 1 − y, −z |
| C4A–H4A⋯O1A | 0.93 | 2.48 | 2.970(3) | 113 | 1 − x, y, 1/2 − z |
| C4B–H4B⋯O1B | 0.93 | 2.43 | 2.905(3) | 111 | x, y, z |
| C6A–H6A⋯O2A | 0.93 | 2.63 | 3.321(3) | 130 | 1 − x, 1 + y, 1/2 − z |
| C6B–H6B⋯O1B | 0.93 | 2.63 | 3.051(3) | 175 | 1/2 − x, 3/2 − y, −z |
| Compound | Cg(1)⋯Cg(2) | Cg(1)a⋯Cg(2)/Å | α b/° | β c | Cg(1)···plane[Cg(2)]/Å | Offsetd/Å | Symm. op. on Cg(2) |
|---|---|---|---|---|---|---|---|
| a Cg = centre of gravity of the aromatic ring. b α = angle between planes of two interacting rings. c β = angle between Cg⋯Cg line and normal to the plane of the first interacting ring. d Offset can be calculated only for the strictly parallel rings (α = 0.00°). For slightly inclined rings (α ≤ 5°) an approximate value is given. | |||||||
| 1 | C1 → C6⋯C1 → C6 | 3.6365(17) | 0.04(13) | 24.2 | 3.3173(11) | 1.490 | −x, 1 − y, 1 − z |
| N1 → C9⋯N1 → C9 | 3.635(2) | 0.0(2) | 26.0 | 3.2668(15) | 1.594 | 1 − x, −y, 2 − z | |
| 2 | Cu1 → O2⋯Cu1 → O4 | 3.6526(11) | 6.49(8) | 22.82 | 3.2181(7) | — | −x, 2 − y, 1 − z |
| Cu1 → O1⋯C4 → C9 | 3.6244(10) | 2.59(8) | 26.91 | 3.2777(7) | 1.64d | −x, 2 − y, 1 − z | |
| Cu1 → O4⋯Cu1 → O4 | 4.0643(10) | 0 | 33.74 | 3.3798(7) | 2.257 | −x, 2 − y, 1 − z | |
| C3 → C3i⋯C4 → C9 | 3.5689(11) | 0.82(8) | 24.09 | 3.2370(7) | 1.46d | 1 + x, y, z | |
| C4 → C9⋯Cu1 → O2 | 3.6244(10) | 2.59(8) | 25.27 | 3.2320(7) | 1.55d | −x, 2 − y, 1 − z | |
| C4 → C9⋯C3 → C3i | 3.5690(11) | 0.82(8) | 24.91 | 3.2582(7) | 1.50d | −1 + x, y, z | |
| 3 | Cu1 → O2⋯Cu1 → O4ii | 3.6885(17) | 6.55(13) | 26.27 | 3.1566(12) | — | −x, 1 − y, 1 − z |
| Cu1 → O2⋯C1 → C6 | 3.7498(18) | 2.60(14) | 31.57 | 3.2735(12) | 1.63d | 1 − x, 1 − y, 1 − z | |
| C1 → C6⋯C1 → C6 | 3.6670(19) | 0.00 | 26.91 | 3.2700(13) | 1.660 | 1 − x, 1 − y, 1 − z | |
| 6 | N1A → C6A⋯C1B → C3B | 4.0151(15) | 2.24(15) | 33.3 | 3.2684(10) | 2.22d | 1 − x, y, 1/2 − z |
Packing of 2 reveals a 3D network realised through seven symmetry-independent hydrogen bonds (Table 3). Complex anions [Cu2(CA)3(H2O)2]2− and uncoordinated water molecules form a porous honeycomb-like structure (Fig. 4a) which is further stabilised by π-stacking of the anions in the direction [100] (Fig. 4b, Table 4). Cations of 4,4′-bpy fill in the voids (Fig. 4a) and they are linked into water–anion network by N–H⋯O hydrogen bonds (Table 3). The rationale behind use of 4,4′-bpy was that it may occupy apical positions in the Cu coordination sphere, linking the linear polymers into the layers, similar to the previously known pyrazine complex, [Cu(CA)(pyz)]n.13
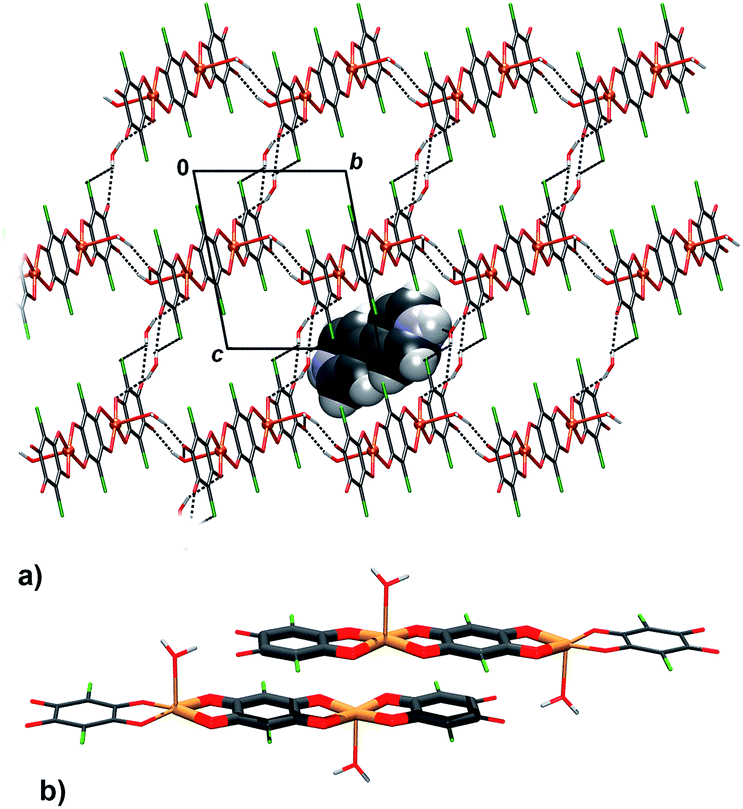 | ||
| Fig. 4 Crystal packing of 2: (a) the O–H⋯O hydrogen-bonded (dashed lines) honeycomb-like network encapsulating 4,4′-bpy cations, (b) π-stacking of planar, delocalised quinoid rings and chelate rings from two [Cu2(CA)3(H2O)2]2− anions (rings participating in the interaction are highlighted). | ||
However, basicity of 4,4′-bpy is too high, so it gets protonated in the presence of moderately strong chloranilic acid (its pKa values being 0.76 and 3.08, respectively47), therefore it is present in the compound in the cationic state. Attempts of moderating pH of the aqueous medium by use of a weak, non-complexing base such as ammonia, yielded no single crystals suitable for X-ray measurement, and the obtained samples were too inhomogeneous and impure to allow any meaningful analysis. Due to the presence of the 4,4′-bpy cations, the infinite neutral [Cu(CA)]n chains (as present in studied compounds 3–6) did not form; instead, discrete dianionic complexes, balancing the charge of the 4,4′-bpy cations, were obtained. However, it is worth nothing that the anions [Cu(CA)2(H2O)2]2− and [Cu2(CA)3(H2O)2]2− are actually fragments of the polymeric chains; thus, we may speculate that the [Cu(CA)]n polymer is generated by addition of similar discrete [Cu(CA)2] or [Cu2(CA)3] units. In the case of 1 and 2 growth of chains was probably terminated due to the presence of the cations.
Infinite chains of 3 extend in the direction [100] (Fig. 5a); the square-pyramidal coordination of Cu centres allows close contact between planar delocalised quinoid and chelate rings, so two chains stack into pairs by π-interactions (Fig. 5b, Table 4). An electron delocalization occurring in the five-membered Cu-chelate rings make them susceptible to interactions with other π-systems in the unit cell;48–52 in fact, it appears that they are quite common in metal–organic chelate complexes.26,27,48–52 There are also C–H⋯O hydrogen bonds which link the pairs of chains into layers parallel to (011) (Fig. 5a, Table 3).
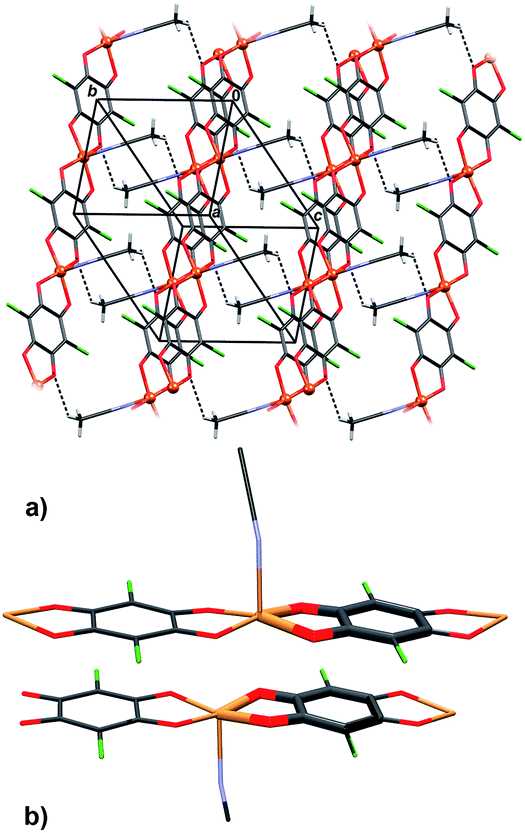 | ||
| Fig. 5 Crystal packing of 3: (a) an arrangement of polymeric chains linked by C–H⋯O hydrogen bonds (dashed lines), (b) showing pairs of π-stacked rings of quinoid ligands and chelate rings (interactions between individual rings are highlighted). | ||
In crystal packing of 4 and 5 linear 1D coordination polymers are extended in the directions [100] (Fig. 6 and 7). Since octahedral coordination of Cu centres prevents close contact between π-systems, there are no stacking interactions and crystal packing is realised through O–H⋯O and O–H⋯Cl hydrogen bonding. Due to steric similarities and existence of inter-chain hydrogen bonding (there are two symmetry-independent hydrogen bonds in both compounds, Table 3), the comparison of their crystal packing (Fig. 6 and 7) reveals inter chain hydrogen bonds in 4 generate a 3D hydrogen-bonding network, while in 5 there are hydrogen-bonded layers. The previously published hydrate of 4, {[Cu(CA) (H2O)2]·H2O}n,31 also forms a 3D network; its packing includes uncoordinated water molecule linking the chains, rather than direct hydrogen bonding between the chains.
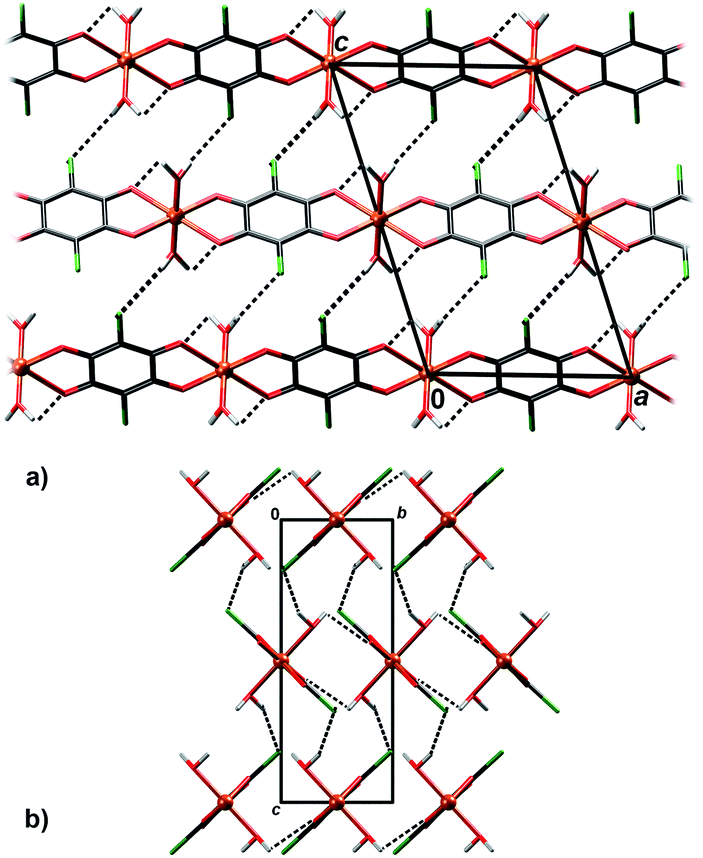 | ||
| Fig. 6 Crystal packing of 4 showing hydrogen bonding O–H⋯Cl and O–H⋯O (dashed lines) between the polymeric chains: (a) viewed in the direction [010], (b) in the direction [100]. | ||
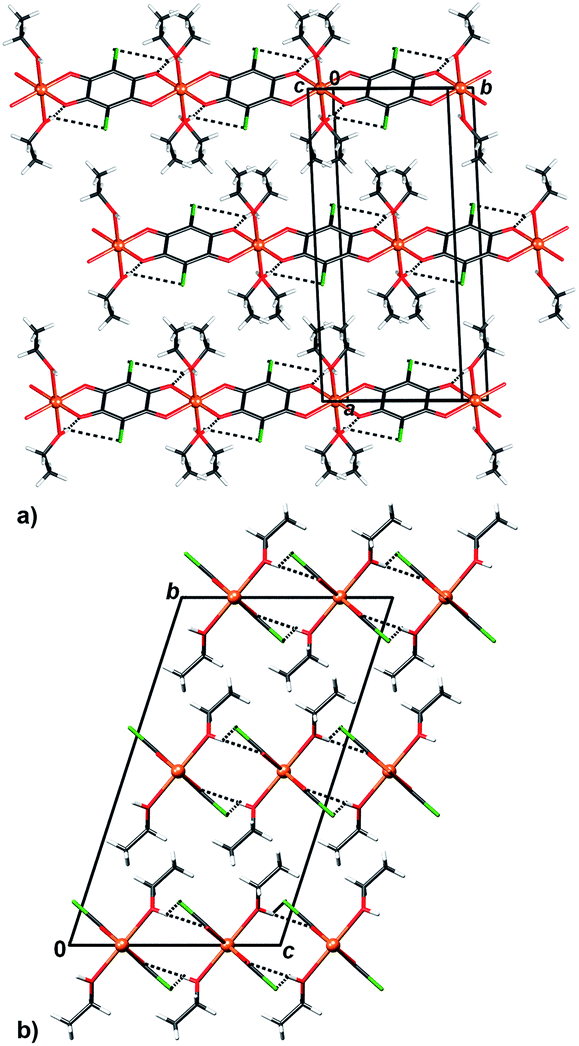 | ||
| Fig. 7 Crystal packing of 5 showing inter chain (chains running parallel to [100]) hydrogen bonding O–H⋯O and O–H⋯Cl (dashed lines) viewed in the direction (a) approximately [0 −2 1] and (b) [100]. | ||
Crystal packing of 6 is generally similar to packing of 4 and 5: polymeric chains are hydrogen bonded into a 3D network (Fig. 8, Table 3). Imidazole ligands of A chains are proton donors towards chloranilate oxygens of B chains and vice versa, forming layers parallel to (110); additionally the layers are stabilised by π-interactions between imidazole ligands (Fig. 8, Table 4). A 3D network is achieved by linking the layers through N–H⋯O hydrogen bonds (Table 3).
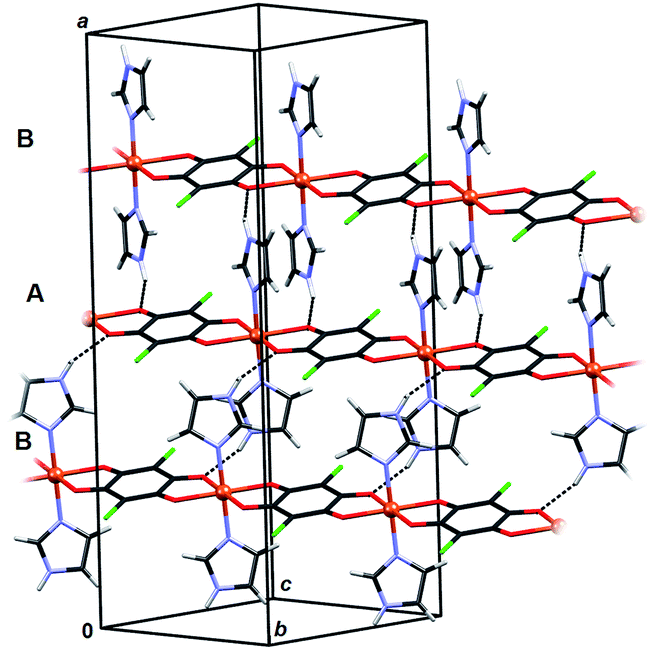 | ||
| Fig. 8 Crystal packing of 6 showing hydrogen bonding N–H⋯O (dashed lines) between the polymeric chains and π-interactions between imidazole ligands. Chains A (C2-symmetric) and B (centrosymmetric) are marked. | ||
ESR spectroscopy
The electron spin resonance (ESR) spectra were recorded from liquid nitrogen temperature up to room temperature (RT) and the representative spectra, obtained at T = 80 and 296 K, are shown in Fig. 9. The relative intensities of the spectra at low and room temperatures are presented in the real ratios, for each compound. Half-field ESR line connected with forbidden transition ΔMs = ±2 was not detected for any compound. To determine spin-Hamiltonian parameters of Cu2+ ions and exchange interaction parameters, spectra simulation and linewidth consideration were performed.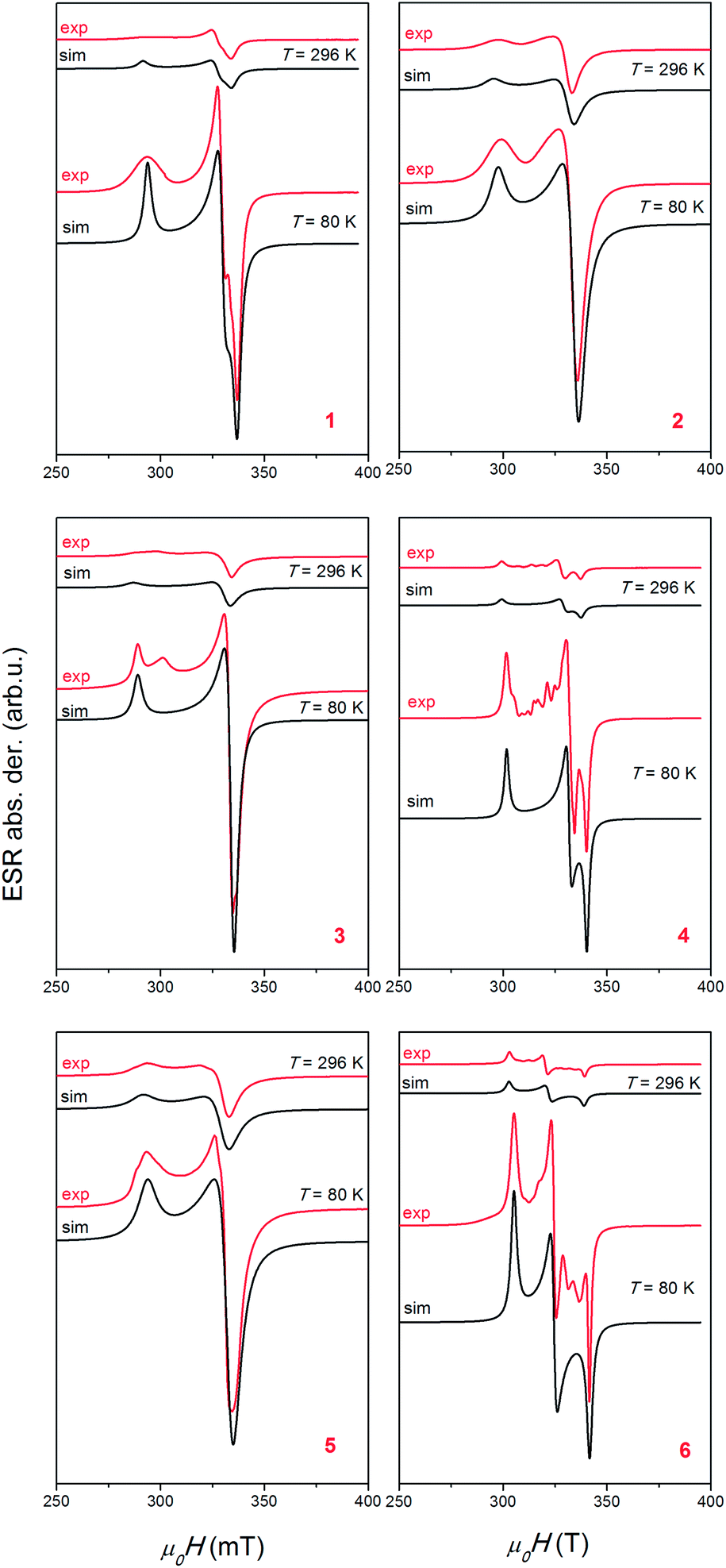 | ||
| Fig. 9 Experimental (red lines) and simulated (black lines) ESR spectra of polycrystalline samples of compounds (Him)2[Cu(CA)2(H2O)2] (1), (4,4′-H2bpy)[Cu2(CA)3(H2O)2]·2H2O (2), [Cu(CA)(CH3CN)]n (3), [Cu(CA)(H2O)2]n (4), [Cu(CA)(EtOH)2]n (5) and [Cu(CA)(im)2]n (6). The intensities of the spectra at T = 80 and 296 K are presented in the real ratios, for each compound. | ||
The simulations were carried out by the EasySpin software.53 To simulate spectra, the following form of spin-Hamiltonian was assumed:
| H = μBB·g·S | (1) |
In eqn (1), B is magnetic field, S is electron spin operator and g is g-tensor. Hyperfine splitting tensor A, due to interaction between electron S = 1/2 and nuclear spin I = 3/2 in copper ions, is approximated to be zero. The simulated spectra, shown in Fig. 9, were obtained using the parameters presented in Table 5. The same parameters were used for the simulation of the spectra at low and room temperatures by taking into consideration only line-broadening effect (ESR lines were broader at higher temperatures). Line shapes used in simulation were always Lorentzian. The simulations reproduce well the experimentally observed spectra for compounds 1, 2 and 5, as could be seen in Fig. 9. The additional lines observed in spectra of compounds 3, 4 and 6 are probably connected with presence of trace impurity (i.e. electron in the vicinity of 14N and 1H nuclei).
| Compound | 1 | 2 | 3 | 4 | 5 | 6 |
| g x | 2.058 | 2.07 | 2.070 | 2.039 | 2.08 | 2.035 |
| g y | 2.103 | 2.07 | 2.078 | 2.092 | 2.08 | 2.143 |
| g z | 2.360 | 2.33 | 2.40 | 2.30 | 2.36 | 2.278 |
| R | 0.175 | 0 | 0.02 | 0.25 | 0 | 0.8 |
| |J| (cm−1) | 0.03 | 0.27 | 0.52 | 0.66 | 0.10 | 0.13 |
Observed Lorentzian lineshapes and absence of hyperfine interaction for copper(II) ions point to the presence of exchange interactions in the investigated systems. The values of exchange interaction parameters, |J|, could be approximately calculated using linewidth analysis and the method of moments:54,55
| Γexp ∼ γ(Γd)2/ωexch | (2) |
Here, ωexch ∼ J[S(S + 1)]1/2 and γ is gyromagnetic ratio, Γexp is the experimental linewidth (Γ = FWHM/(2 × 1.18)) while Γd is the dipolar linewidth due to the contribution of the nearest copper(II) ions.56,57 If we consider all copper ions in the sphere of radius 15 Å, the values of the dipolar linewidths are Γd ∼ 5–25 mT. Taking into consideration experimental linewidths (values at 80 K used in the simulations) Γexp ∼ 0.7–2.6 mT, exchange interaction parameters, |J|, were calculated and presented in Table 5. The values calculated in this way have approximate accuracy, but from Table 5 it could be seen that weak exchange interaction are found for all compounds (|J| < 1 cm−1).
The obtained axial g-tensors with gz > gx = gx reveal that the ground state of unpaired electron is the dx2−y2 orbital, for compounds 2 and 5. Other compounds show “rhombic” spectra, exhibiting three different g-values. For complexes of this type, a parameter R is the indication of the predominance of the dx2−y2 or dz2 orbital in the ground state.58 The systems where gz > gy > gx, R parameter is defined as:
| R = (gy − gx)/(gz − gy) | (3) |
For R > 1, the ground state is predominantly dz2, whereas for R < 1, the ground state is predominantly dx2−y2. The obtained R parameters are shown in Table 5, indicating that the greater contribution to the ground state arises from dx2−y2 orbital. This is in agreement with the crystal structure of the compounds. Both types of observed coordination of copper atoms (a square pyramid in compounds 2 and 3 and elongated octahedron in compounds 1, 4, 5 and 6) give so called normal spectra of Cu2+ ions (gz > gy ≈ gx > 2.0023).58
The distance between chloranilate-bridged Cu2+ atoms within a single molecule range between 7.6 and 8.1 Å, while the inter chain distances of the Cu2+ atoms are considerably shorter than intrachain ones (Table 6). However, relative orientations of dx2−y2 orbitals of interacting Cu2+ atoms are important. In 1, 2, 3 and 5, the dx2−y2 orbital is parallel to the chloranilate plane, while in 4 and 6 it lies in the plane defined by Cu–O2(CA) and Cu–Laxial vectors, approximately normal to the chloranilate plane9,10 (Table 1). However, in all studied compounds, orientations of dx2−y2 orbitals in neighbouring molecules are not favourable for orbital overlap.57 Therefore, weak exchange interaction is observed inside [Cu(CA)]n chains.
| Compound | No. of close contacts | Close intrachain distance/Å | Close interchain distance/Å |
|---|---|---|---|
| 1 | 2 | — | 8.747 |
| 2 | 1 | 7.735 | 3.877 |
| 3 | 1 | 7.578 | 3.937 |
| 4 | 2 | 8.087 | 4.832 |
| 5 | 1 | 7.650 | 5.199 |
| 6 | 2 | 8.107 | 6.929 |
Conclusions
We have prepared and characterised six novel Cu–chloranilate compounds, ranging from mononuclear to infinite 1D coordination polymers. Due to small size of the Cu2+ atom, it can accommodate two chloranilate ligands in trans-arrangement, only; according to the Cambridge Structural Database,59 for larger first-row transition metals the arrangement is usually cis (generating zig–zag coordination polymers). The fifth and sixth coordination sites can then be occupied by small monodentate ligands such as water, ethanol or acetonitrile, which do not introduce steric strain; therefore, [Cu(CA)]n coordination polymers remain linear. A bidentate ancillary ligand, such as 2,2′-bipyridine, must bind cis vs. chloranilate; in the case of the small Cu2+ atom, it is impossible, and monomers are obtained, only.26,27 However, the bulky aromatic imidazole ligand serendipitously revealed interesting steric effects: instead of bridging, it binds to the linear [Cu(CA)]n chains as a monodentate ligand; its steric strain is compensated by its different orientations, resulting in a combination of chiral (A) and achiral meso-(B) chains. Also, cross-linking the [Cu(CA)]n polymers into a 2D network is not achieved easily, since both imidazole and 4,4′-bipyridine are highly basic and tend to be protonated in the presence of chloranilic acid. Raising pH of the solution by use of some common bases usually results in unwanted precipitation of alkali or ammonium chloranilate salts and/or copper hydroxyde.In coordination polymers, copper(II) usually has a distorted octahedral coordination, and the compound 3 is a rare example of Cu–chloranilate coordination polymer with pentacoordinated Cu2+. Also, compound 2 is a rare example of a complex with two different modes of coordination of chloranilate, a terminal bidentate and a bridging (bis)bidentate ones. Changes of electronic structure of the 2,5-dihydroxyquinonate ring upon various coordination modes affects significantly geometry of the chloranilate moiety.
To the best of our knowledge, so far no crystal structure comprising both a racemate and a meso-compound has been described. The compound 6 is probably the first such example.
ESR spectroscopy gave the g-tensors for Cu(II) ions. The spectra simulations reveal dx2−y2 orbital as the ground state of unpaired electron in copper ions for compounds 2 and 5, whereas for other four compounds, due to “rhombic” spectra, the greater contribution to the ground state arises from dx2−y2 orbital. These results are in agreement with cooper coordination, which is square pyramid in the compounds 2 and 3, and elongated octahedron in other compounds. Furthermore, ESR linewidth analysis revealed the presence of weak exchange interactions (|J| < 1 cm−1) between the copper ions in the investigated complexes, which is consistent with specific crystal packing and also with the values found in similar compounds.9,10
Experimental
Materials and physical measurements
The chemicals were purchased from commercial sources, and used without further purification. Infrared spectra were recorded as KBr pellets using a Bruker Alpha-T spectrometer, in the 4000–350 cm−1 range.Preparation of compounds 1–6
IR data (KBr, cm−1): 3434 (m, br), 3153 (w), 2923 (w), 2852(w), 1657 (w), 1640 (s), 1597 (m), 1535 (s), 1499 (m), 1462 (vs), 1350 (w), 1340 (m), 1269 (w), 1203 (w), 1177 (sh), 1102 (w), 1059 (m), 1003 (w), 944 (w), 917 (w), 851 (m), 825 (w), 781 (w), 722 (w), 669 (w), 622 (w), 611 (w), 569 (m), 541 (w), 515 (w), 446 (w).
IR data (KBr, cm−1): 3424 (m, br), 3130 (w), 3084 (w), 2922 (w), 2852(w), 1637 (m), 1609 (m), 1588 (w), 1545 (s), 1508 (vs), 1366 (vs), 1337 (m), 1266 (w), 1242 (w), 1203 (w), 1131 (w), 1107 (w), 1036 (w), 1001 (w), 866 (m), 847 (m), 797 (m), 710 (w), 626 (w), 599 (w), 573 (m), 527 (w), 461 (w), 414 (w).
IR data (KBr, cm−1): 2923 (w), 2852 (w), 1707 (w), 1620 (w), 1490 (vs), 1370 (vs), 1314 (m), 1285 (w), 1092 (sh), 1011 (w), 866 (s), 635 (w), 579 (m), 537 (m), 517 (sh), 469 (w).
IR data (KBr, cm−1): 3432 (br, m), 2922 (w), 2858 (m), 1709 (w), 1617 (m), 1507 (vs), 1380 (s), 1290 (w), 1256 (w), 1006 (w), 959 (w), 856 (m), 805 (w), 698 (w), 603 (w), 579 (m), 481 (w), 401 (w).
IR data (KBr, cm−1): 3451 (m, br), 2922 (w), 2852 (w), 1705 (w), 1620 (w), 1486 (vs), 1368 (vs), 1314 (m), 1093 (w), 1051 (sh), 1011 (w), 866 (s), 635 (w), 580 (m), 540 (m), 516 (w), 468 (w).
IR data (KBr, cm−1): 2924 (w), 2853 (w), 1627 (s), 1512 (vs), 1437 (w), 1370 (m), 1328 (w), 1293 (w), 1257 (w), 1204 (w), 1179 (w), 1132 (w), 1101 (w), 1070 (m), 1004 (w), 994 (w), 948 (w), 919 (w), 851 (m), 826 (w), 768 (w), 672 (w), 656 (w), 618 (w), 600 (w), 576 (m), 541 (w), 517 (w), 465 (w).
Crystallographic data collection and refinement
Single crystal measurements were performed on an Oxford Diffraction Xcalibur Nova R diffractometer (microfocus Cu tube) at RT [293(2) K]. Only the symmetry-independent part of the Ewald sphere was measured. Program package CrysAlis PRO60 was used for data reduction. The structures were solved using SHELXS97 (ref. 61) and refined with SHELXL97.61 The models were refined using the full-matrix least squares refinement; all non-hydrogen atoms were refined anisotropically. Hydrogen atoms bound to C atoms were modelled as riding entities using the AFIX command, while those bound to O were located in difference Fourier maps and refined with the following restraints: geometry of water molecules was restrained to d(O–H) = 0.95(2) Å; d(H⋯H) = 1.50(4) Å, and the hydroxyl group to d(O–H) = 0.82(2) Å. In 2 methyl hydrogen were refines as free atoms with the following restraints: d(C–H) = 0.96(2) Å; d(H⋯H) = 1.50(4) Å.Molecular geometry calculations were performed by PLATON,62 and molecular graphics were prepared using ORTEP-3,33 and CCDC-Mercury.63 Crystallographic and refinement data for the structures reported in this paper are shown in Table 7.
| Compound | 1 | 2 | 3 | 4 | 5 | 6 |
| Empirical formula | C18H14Cl4CuN4O10 | C28H18Cl6Cu2N2O16 | C8H3Cl2CuNO4 | C6H4Cl2CuO6 | C10H12Cl2CuO6 | C12H8Cl2CuN4O4 |
| Formula wt/g mol−1 | 651.68 | 978.24 | 311.55 | 306.53 | 362.64 | 406.67 |
| Crystal dimensions/mm | 0.15 × 0.12 × 0.10 | 0.12 × 0.10 × 0.05 | 0.09 × 0.07 × 0.04 | 0.10 × 0.05 × 0.05 | 0.20 × 0.16 × 0.12 | 0.20 × 0.04 × 0.04 |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/c | P21/c | C2/c | C2/c |
| a/Å | 8.2635(10) | 9.3314(5) | 7.5779(3) | 8.0866(6) | 17.8431(10) | 25.3992(8) |
| b/Å | 8.7473(8) | 9.4880(4) | 8.2261(4) | 4.8324(4) | 7.6498(4) | 8.1066(2) |
| c/Å | 9.1791(7) | 10.3517(6) | 16.7585(8) | 12.9148(9) | 10.3003(5) | 14.2791(4) |
| α/° | 70.332(8) | 80.069(4) | 90 | 90 | 90 | 90 |
| β/° | 71.570(9) | 89.845(4) | 94.337(4) | 108.388(8) | 107.903(6) | 96.491(3) |
| γ/° | 67.319(10) | 66.524(5) | 90 | 90 | 90 | 90 |
| Z | 1 | 1 | 4 | 2 | 4 | 8 |
| V/Å3 | 563.05(10) | 825.83(7) | 1041.68(8) | 478.91(6) | 1337.87(12) | 2921.24(14) |
| D calc/g cm−3 | 1.922 | 1.967 | 1.987 | 2.126 | 1.800 | 1.849 |
| μ/mm−1 | 6.356 | 6.795 | 7.703 | 8.484 | 6.183 | 5.735 |
| θ range/° | 5.24–75.99 | 4.35–75.85 | 5.29–75.69 | 3.61–75.81 | 6.35–75.75 | 3.50–75.92 |
| T/K | 293(2) | 293(2) | 293(2) | 293(2) | 293(2) | 293(2) |
| Radiation wavelength | 1.54179 (CuKα) | 1.54179 (CuKα) | 1.54179 (CuKα) | 1.54179 (CuKα) | 1.54179 (CuKα) | 1.54179 (CuKα) |
| Diffractometer type | Xcalibur Nova | Xcalibur Nova | Xcalibur Nova | Xcalibur Nova | Xcalibur Nova | Xcalibur Nova |
| Range of h, k, l | −9 < h < 10 | −11 < h < 11 | −9 < h < 9 | −10 < h < 10 | −22 < h < 21 | −31 < h < 29 |
| −9 < k < 10 | −8 < k < 12 | −10 < k < 6 | −6 < k < 6 | −9 < k < 8 | −10 < k < 6 | |
| −7 < l < 11 | −12 < l < 12 | −21 < l < 20 | −16 < l < 10 | −12 < l < 8 | −17 < l < 17 | |
| Reflections collected | 4503 | 6802 | 4736 | 4605 | 2868 | 6948 |
| Independent reflections | 2278 | 3371 | 2146 | 1047 | 1367 | 2994 |
| Observed reflections (I ≥ 2σ) | 2094 | 2993 | 1888 | 960 | 1056 | 2373 |
| Absorption correction | Multi-scan | Multi-scan | Multi-scan | Multi-scan | Multi-scan | Multi-scan |
| R int | 0.0400 | 0.0224 | 0.0361 | 0.0439 | 0.0370 | 0.0275 |
| R(F) | 0.0429 | 0.0356 | 0.0444 | 0.0730 | 0.0479 | 0.0353 |
| R w(F2) | 0.1270 | 0.1053 | 0.1292 | 0.1913 | 0.1452 | 0.1075 |
| Goodness of fit | 1.077 | 1.048 | 1.060 | 1.144 | 1.146 | 1.028 |
| H atom treatment | Mixed | Mixed | Free | Free | Mixed | Riding |
| No. of parameters | 177 | 260 | 157 | 79 | 88 | 210 |
| No. of restraints | 3 | 6 | 9 | 3 | 1 | 0 |
| Δρmax, Δρmin (eÅ−3) | 0.528; −0.751 | 0.476; −0.502 | 0.490; −0.801 | 0.561; −0.592 | 0.452; −0.708 | 0.376; −0.508 |
ESR spectroscopy
The ESR measurements were performed on polycrystalline samples of the compounds 1–6 by an X-band Bruker Elexsys 580 FT/CW spectrometer (microwave frequency around 9.7 GHz). The measurements were carried out at the modulation frequency 100 kHz. The magnetic field modulation amplitude was 0.5 mT (recorded also with modulation amplitude of 0.2 mT, as additional verification). The spectrometer was equipped with a standard Oxford Instruments model DTC2 temperature controller. Spectra were recorded from liquid nitrogen temperature up to RT (Fig. 9).Acknowledgements
Financial support by the Croatian Science Foundation (grants no. IP-2014-09-4079 and 1108) and also by the Croatian Academy of Sciences and Arts (grant for 2014) is gratefully acknowledged.Notes and references
- S. L. James, Chem. Soc. Rev., 2003, 32, 276–288 RSC.
- T. Yamada, K. Otsubo, R. Makiura and H. Kitagawa, Chem. Soc. Rev., 2013, 42, 6655–6669 RSC.
- C. Janiak, Dalton Trans., 2003, 2781–2804 RSC.
- C. Janiak and J. K. Vieth, New J. Chem., 2010, 34, 2366–2388 RSC.
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- N. Hoshino, F. Ijima, G. N. Newton, N. Yoshida, T. Shiga, H. Nojiri, A. Nakao, R. Kumai, Y. Murakami and H. Oshio, Nat. Chem., 2012, 4, 921–926 CrossRef CAS PubMed.
- E. Koumousi, I.-R. Jeon, Q. Gao, D. Dechambenoit, D. N. Woodruff, P. Merzeau, L. Buisson, X. Jia, D. Li, F. Volatron, C. Mathonière and R. Clérac, J. Am. Chem. Soc., 2014, 136, 15461–15464 CrossRef CAS PubMed.
- L. Cao, Q. Gao, T. Liu, Z. Xia and D. Li, Chem. Commun., 2014, 50, 1665–1667 RSC.
- M. Nihei, Y. Sekine, N. Suganami, K. Nakazawa, A. Nakao, H. Nakao, Y. Murakami and H. Oshio, J. Am. Chem. Soc., 2011, 133, 3592–3600 CrossRef CAS PubMed.
- (a) S. Kitagawa and S. Kawata, Coord. Chem. Rev., 2002, 224, 11–34 CrossRef CAS; (b) S. Kitagawa and R. Matsuda, Coord. Chem. Rev., 2007, 251, 2490–2509 CrossRef CAS.
- (a) S. Kawata, S. Kitagawa, H. Kumagai, C. Kudo, H. Kamesaki, T. Ishiyama, R. Suzuki, M. Kondo and M. Katada, Inorg. Chem., 1996, 35, 4449–4461 CrossRef CAS PubMed; (b) C. N. Rao, S. Natarajan and R. Vaidhyanathan, Angew. Chem., Int. Ed., 2004, 43, 1466–1496 CrossRef CAS PubMed; (c) X.-Y. Wang, C. Avendaño and K. R. Dunbar, Chem. Soc. Rev., 2011, 40, 3213–3238 RSC.
- M. K. Kabir, N. Miyazaki, S. Kawata, K. Adachi, H. Kumagai, K. Inoue, S. Kitagawa, K. Ijima and M. Katada, Coord. Chem. Rev., 2000, 198, 157–169 CrossRef CAS.
- S. Kawata, S. Kitagawa, M. Kondo, I. Furuchi and M. Munakata, Angew. Chem., Int. Ed., 1994, 33, 1759–1761 CrossRef.
- S. Kawata, S. Kitagawa, H. Kumagai, T. Ishiyama, K. Honda, H. Tobita, K. Adachi and M. Katada, Chem. Mater., 1998, 10, 3902–3912 CrossRef CAS.
- B. F. Abrahams, K. D. Lu, B. Moubaraki, K. S. Murray and R. Robson, J. Chem. Soc., Dalton Trans., 2000, 1793–1797 RSC.
- S. Kawata, H. Kumagai, K. Adachi and S. Kitagawa, J. Chem. Soc., Dalton Trans., 2000, 2409–2417 RSC.
- B. F. Abrahams, J. Coleiro, K. Ha, B. F. Hoskins, S. D. Orchard and R. D. Robson, Dalton Trans., 2002, 1586–1594 RSC.
- B. F. Abrahams, M. J. Grannas, T. A. Hudson, S. A. Hughes, N. H. Pranoto and R. D. Robson, Dalton Trans., 2011, 40, 12224–12247 RSC.
- M. Atzori, S. Benmansour, G. Mínguez Espallargas, M. Clemente-Léon, A. Abhervé, P. Gómez-Claramunt, E. Coronado, F. Artizzu, E. Sessini, P. Deplano, A. Serpe, M. L. Mercuri and C. J. Gómez García, Inorg. Chem., 2013, 52, 10031–10040 CrossRef CAS PubMed.
- M. Atzori, F. Pop, P. Auban-Senzier, R. Clérac, E. Canadell, M. L. Mercuri and N. Avarvari, Inorg. Chem., 2015, 54, 3643–3653 CrossRef CAS PubMed.
- M. Hernández-Molina, P. A. Lorenzo-Luis and C. Ruiz-Pérez, CrystEngComm, 2001, 3, 60–63 RSC.
- B. F. Abrahams, T. A. Hudson, L. J. McCormick and R. Robson, Cryst. Growth Des., 2011, 11, 2717–2720 CAS.
- M. Atzori, F. Artizzu, E. Sessini, L. Marchió, D. Loche, A. Serpe, P. Deplano, G. Concas, F. Pop, N. Avarvari and M. L. Mercuri, Dalton Trans., 2014, 43, 7006–7019 RSC.
- S. Benmansour, C. Vallés-García and C. J. Gómez-García, Structural Chemistry & Crystallography Communication, 2015, 1(1), 2 Search PubMed.
- O. Kahn, Molecular Magnetism, Wiley-VCH, New York, 1993 Search PubMed.
- K. Molčanov, M. Jurić and B. Kojić-Prodić, Dalton Trans., 2014, 43, 7208–7218 RSC.
- K. Molčanov, M. Jurić and B. Kojić-Prodić, Dalton Trans., 2013, 42, 15756–15765 RSC.
- K. Molčanov, B. Kojić-Prodić, D. Babić, D. Žilić and B. Rakvin, CrystEngComm, 2011, 13, 5170–5178 RSC.
- K. Molčanov, D. Babić, B. Kojić-Prodić, J. Stare, N. Maltar-Strmečki and L. Androš, Acta Crystallogr., Sect. B: Struct. Sci., 2014, 70, 181–190 Search PubMed.
- S. R. Batten, S. M. Neville and D. R. Turner, Coordination Polymers, Design, Analysis and Application, RSC Publishing, London, 2009 Search PubMed.
- (a) S. Cueto, H.-P. Straumann, P. Rys, W. Petter, V. Gramlich and F. S. Rys, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1992, C48, 458–460 CrossRef CAS; (b) S. Kawata, S. Kitagawa, M. Kondo and M. Katada, Synth. Met., 1995, 71, 1917–1918 CrossRef CAS.
- B. Spingler, S. Schnidrig, T. Todorova and F. Wild, CrystEngComm, 2012, 14, 751–757 RSC.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- Y.-H. Chung, S.-I. Fang, S.-H. Fang, H.-H. Lin and S.-Y. Liou, J. Chin. Chem. Soc., 2009, 56, 1099–1107 CrossRef CAS.
- C. Fujii, M. Mitsumi, M. Kodera, K.-I. Motoda, M. Ohba, N. Matsumoto and H. Okawa, Polyhedron, 1994, 13, 933–938 CrossRef CAS.
- C. G. Pierpont, L. C. Francesconi and D. N. Hendrickson, Inorg. Chem., 1977, 16, 2367–2376 CrossRef CAS.
- H. Amouri and M. Gruselle, Chirality in Transition Metal Chemistry: Molecules, Supramolecular Assemblies and Materials, Wiley, Chichester, U.K, 2008 Search PubMed.
- M. Kawahara, Md. Khayrul Kabir, K. Yamada, K. Adachi, H. Kumagai, Y. Narumi, K. Kindo, S. Kitagawa and S. Kawata, Inorg. Chem., 2004, 43, 92–100 CrossRef CAS PubMed.
- S. Benmansour, V. Vallés-García, P. Gómez-Claramunt, G. Mínguez Espallargas and C. J. Gómez-García, Inorg. Chem., 2015, 54, 5410–5418 CrossRef CAS PubMed.
- T.-T. Luo, Y.-H. Liu, H.-L. Tsai, C.-C. Su, C.-H. Ueng and K.-L. Lu, Eur. J. Inorg. Chem., 2004, 4253–4258 CrossRef CAS.
- P. C. A. Bruijnincx, M. Viciano-Chumillas, M. Lutz, A. L. Spek, J. Reedijk, G. van Koten and R. J. M. Klein Gebbink, Chem.–Eur. J., 2008, 14, 5567–5576 CrossRef CAS PubMed.
- J. V. Folgado, R. Ibáñez, E. Coronado, D. Beltrán, J. M. Savariault and J. Galy, Inorg. Chem., 1988, 27, 19–26 CrossRef CAS.
- Y.-F. Han, Y.-J. Lin, W.-G. Jia and G.-X. Jin, Organometallics, 2008, 27, 4088–4097 CrossRef CAS.
- N. Biliškov, B. Kojić-Prodić, G. Mali, K. Molčanov and J. Stare, J. Phys. Chem. A, 2011, 115, 3154–3166 CrossRef PubMed.
- A. Pawlukojć, G. Bator, L. Sobczyk, E. Grech and J. Nowicka-Scheibe, J. Phys. Org. Chem., 2003, 16, 709–714 CrossRef.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley & Sons, New York, 6th edn, 2009 Search PubMed.
- K. Molčanov, B. Kojić-Prodić and A. Meden, Croat. Chem. Acta, 2009, 82, 387–396 Search PubMed.
- H. Masui, Coord. Chem. Rev., 2001, 219–221, 957–992 CrossRef CAS.
- M. K. Miličić, B. D. Ostojić and S. D. Zarić, Inorg. Chem., 2007, 46, 7109–7114 CrossRef PubMed.
- M. Pitoňak, P. Neogrády, J. Řezáč, P. Jurečka, M. Urban and P. Hobza, J. Chem. Theory Comput., 2008, 4, 1829–1834 CrossRef PubMed.
- D. N. Sredojević, D. Z. Vojisavljević, Z. D. Tomić and S. Zarić, Acta Crystallogr., Sect. B: Struct. Sci., 2012, 68, 261–265 Search PubMed.
- The Importance of Pi-Interactions in Crystal Engineering, ed. E. R. T. Tiekink and J. Zukerman-Schpector, Wiley & Sons, Inc., New York, 2012 Search PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- C. P. Poole, Electron Spin Resonance, A Comprehensive Treatise an Experimental Techniques, Wiley, New York, 1982 Search PubMed.
- M. Goldman, Spin Temperature and NMR in Solids, Oxford University Press, 1970 Search PubMed.
- L. Androš, M. Jurić, P. Planinić, D. Žilić, B. Rakvin and K. Molčanov, Polyhedron, 2010, 29, 1291–1298 CrossRef.
- D. Žilić, B. Rakvin, D. Milić, D. Pajić, I. Đilović, M. Cametti and Z. Džolić, Dalton Trans., 2014, 43, 11877–11887 RSC.
- E. Garribba and G. Micera, J. Chem. Educ., 2006, 83, 1229–1232 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- CrysAlis PRO, Oxford Diffraction Ltd., U.K., 2007 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. ven de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: CIF files, geometric parameters of copper(II) coordination spheres in compounds 1–6 (Table S1), views of the imidazolium and 4,4′-bipyridinium cations (Fig. S1 and S2). CCDC 1433294–1433297, 1473407 and 1473408. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra13809h |
| This journal is © The Royal Society of Chemistry 2016 |