
Morita–Baylis–Hillman reaction in eutectic solvent under aqueous medium†
Sanhu Zhao*,
Hangyu Zhi,
Mi Zhang,
Qin Yan,
Jianfeng Fan and
Jinchang Guo
Department of Chemistry, Xinzhou Teachers University, Xinzhou 034000, Shanxi, China. E-mail: sanhuzhao@163.com
First published on 20th June 2016
Abstract
A series of deep eutectic solvents (DESs) based on choline chloride were prepared and used in the Morita–Baylis–Hillman (M–B–H) reaction. Research showed that the composite solvent (1ChCl/2Gly DES–H2O) is a very effective solvent media for all substrates tested. Under the mild reaction conditions, DABCO catalyzed M–B–H reactions proceed very quickly and efficiently. It is particularly important that the solid M–B–H adducts suspended in the reaction system at the end of the reaction, which can be obtained by simple filtering. Such simple product processing is not reported in the previous literature. The composite solvent (1ChCl/2Gly DES–H2O) was reused directly without any activation process. After six cycles, the yields almost remained unchanged. This protocol has notable advantages such as low cost, ease of work-up and reuse of DES conveniently, which could contribute to the development of new solvent system for use in continuous chemical processes.
Introduction
The Morita–Baylis–Hillman (M–B–H) reaction, typically catalyzed by tertiary amine bases such as DABCO,1 DBU2 and quinuclidines,3 is one of the most versatile carbon–carbon bond-forming reactions in modern organic synthesis.4 In the past decade, due to its atom economy, mild reaction conditions and generality of functional groups, the M–B–H reaction has attracted much attention.5 However, even for the most favorable system, this reaction often suffers from poor reaction rates and long reaction time. To circumvent this sluggish nature of the reaction, recent efforts in this area have been focused largely on developing efficient reaction systems, which involving using nonionic surfactant Triton X-100,6 aqueous cationic micellar solution,7 ionic liquids,8 PEG-400,9 sulpholane,10 supercritical CO2,11 bio-based solvents,12 aqueous buffer,13 aqueous medium14 and neat conditions.15 However, so far, almost all of the M–B–H products isolation is difficult and the column chromatography was often used to purify the product, which makes its widely application very inconvenient.Recently, deep eutectic solvents (DESs), which have similar physical properties and phase behavior to ILs, are gaining increasing attention in chemistry fields.16 Because of their less toxic and biodegradable properties,17 they have been applied in many fields such as biocatalysis, extraction, carbon dioxide capture, biomedical applications and organic synthesis.18 However, their application in the M–B–H reaction as solvent has not been reported. In our laboratories, we have been committed to developing green solvents that can be used for the organic reactions.19 During our own investigations on the DESs, we have recently found that the choline chloride–glycerol DES qualifies as an efficient, recyclable reaction medium for the Morita–Baylis–Hillman reaction. Because the M–B–H product does not dissolve in the aqueous choline chloride–glycerol DES, the product isolation is very easy and the target product can be isolated by simple filter. Herein, we would like to present the catalytic application of the aqueous choline chloride–glycerol DES in the M–B–H reaction. In comparison to other reported M–B–H reaction systems, the present solvent-catalyst system composed of choline chloride–glycerol DES, H2O and DABCO works very well at room temperature. Furthermore, the use of the solvent system 1ChCl/2Gly DES–H2O as reaction media allows a very efficient catalytic recycling (up to six consecutive times) and simple product isolation.
Results and discussion
Firstly, a series of DESs containing choline chloride were prepared by mixing choline chloride with urea or glycerol at molar ratios of choline chloride to urea or glycerol from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:3 at 85–95 °C for 6–10 h. Then these DESs were used in the M–B–H reaction of 4-chloro-benzaldehyde with acrylonitrile. Experimental results are summarized in Table 1. To our delight, in the presence of DESs containing choline chloride, the M–B–H reaction proceeded quite smoothly and showed a significant acceleration effect with DABCO as base (Table 1, entries 1–5). In contrast, the reaction in common molecular solvents, such as acetonitrile and tetrahydrofuran (THF), gave 30 and 39% yields of the desired M–B–H product after 2880 min (Table 1, entries 7 and 8). While the same reaction in 1ChCl/1Lac DES, only trace amount of product was found after 1440 min (Table 1, entry 6). It was also shown that the ChCl/Gly had a better effect than ChCl/urea (Table 1, entries 1–5). To further confirm the enhancement is derived from DESs itself or individual, the choline chloride and glycerol were separately used as reaction media (Table 1, entries 9 and 10), only 48% and 51% yields of the desired M–B–H product were respectively obtained after 1440 min, so the acceleration is indeed resulted from the DES itself. Among all six DESs containing choline chloride examined, the 1ChCl/2Gly DES provided slightly better results in terms of reaction yield and reaction time (Table 1, entry 4).
:1 to 1:3 at 85–95 °C for 6–10 h. Then these DESs were used in the M–B–H reaction of 4-chloro-benzaldehyde with acrylonitrile. Experimental results are summarized in Table 1. To our delight, in the presence of DESs containing choline chloride, the M–B–H reaction proceeded quite smoothly and showed a significant acceleration effect with DABCO as base (Table 1, entries 1–5). In contrast, the reaction in common molecular solvents, such as acetonitrile and tetrahydrofuran (THF), gave 30 and 39% yields of the desired M–B–H product after 2880 min (Table 1, entries 7 and 8). While the same reaction in 1ChCl/1Lac DES, only trace amount of product was found after 1440 min (Table 1, entry 6). It was also shown that the ChCl/Gly had a better effect than ChCl/urea (Table 1, entries 1–5). To further confirm the enhancement is derived from DESs itself or individual, the choline chloride and glycerol were separately used as reaction media (Table 1, entries 9 and 10), only 48% and 51% yields of the desired M–B–H product were respectively obtained after 1440 min, so the acceleration is indeed resulted from the DES itself. Among all six DESs containing choline chloride examined, the 1ChCl/2Gly DES provided slightly better results in terms of reaction yield and reaction time (Table 1, entry 4).
|
|||
|---|---|---|---|
| Entry | Solvents | Time/min | Yield (%) |
| a General reaction conditions: 4-chlorobenzaldehyde (10 mmol), DABCO (10 mmol), solvent (3.0 g), acrylonitrile (12 mmol), no further increase in yield after the reported time.b Only trace amount of product was detected and its yield not determined.c Under ultrasound irradiation, the ultrasonic power 360 W, irradiation frequency 40 kHz. | |||
| 1 | 1ChCl/1urea | 135 | 94 |
| 2 | 1ChCl/2urea | 120 | 95 |
| 3 | 1ChCl/1Gly | 120 | 97 |
| 4 | 1ChCl/2Gly | 100 | 98 |
| 5 | 1ChCl/3Gly | 130 | 97 |
| 6 | 1ChCl/1Lac | 1440 | NDb |
| 7 | CH3CN | 2880 | 30 |
| 8 | THF | 2880 | 39 |
| 9 | 3 g ChCl | 1440 | 48 |
| 10 | 3 g glycerol | 1440 | 51 |
| 11 | 3 g ChCl +3 g H2O | 1440 | 72 |
| 12 | 3 g 1ChCl/2Gly + 0.5 g H2O | 100 | 98 |
| 13 | 3 g 1ChCl/2Gly + 1.0 g H2O | 80 | 98 |
| 14 | 3 g 1ChCl/2Gly + 1.5 g H2O | 95 | 98 |
| 15 | 3 g H2O | 1440 | NDb |
| 16 | Neat | 1440 | 86 |
| 17c | 3 g 1ChCl/2Gly + 1.0 g H2O | 80 | 98 |
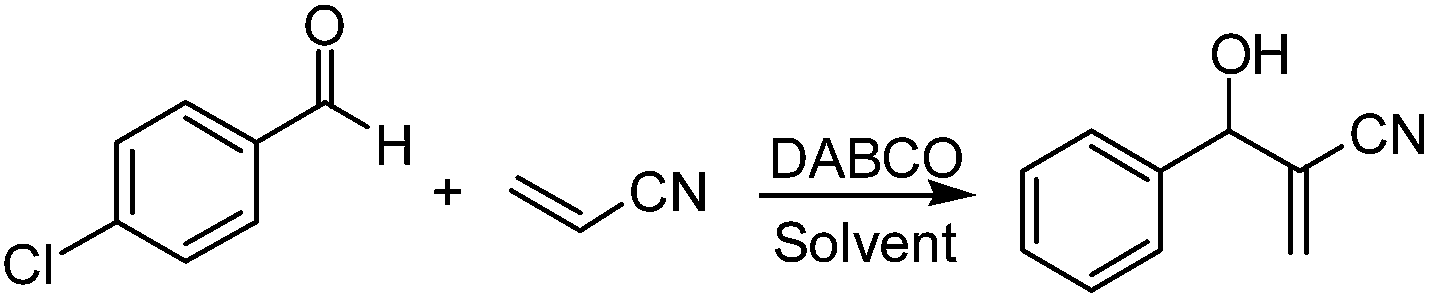
Recently, as a kind of environmental friendly solvent, water has been often used to promote organic reaction.20 To our knowledge, it is generally easy to dissolve DESs in water to form homogeneous system. Comparing the pure DESs or water, the homogeneous system made up of water and DESs may be having more excellent properties. So the 1ChCl/2Gly DES and water were mixed at mass ratios of DES to water from 3:0.5 to 3:1.5, then the mixtures were used as reaction medium in the M–B–H reaction of 4-chloro-benzaldehyde with acrylonitrile (Table 1, entries 12–14). In the presence of water–1ChCl/2Gly composite system, the DABCO catalyzed M–B–H reaction proceeded quite smoothly and showed a significant acceleration effect, when the quality of the water percentage reaches 25% (Table 1, entry 13), the optimal reaction results (yield 98%, 80 min) is got, and most importantly, after the reaction, the product suspended in the reaction system can be obtained by simple filtering. Such simple product processing is not reported in previous literature. When using water alone as solvent, almost no product was detected (Table 1, entry 15). These results indicated that the 1ChCl/2Gly DES–H2O system played a critical role in promoting the M–B–H reaction. Interestingly, when the M–B–H reaction was performed in neat conditions, the 86% yield of desired product was achieved after 1440 min (Table 1, entry 16). Ultrasound-accelerated chemical reactions are well-known and proceed via the formation and adiabatic collapse of the transient cavitation bubbles.21 In order to further optimize the reaction conditions, the ultrasonic irradiation was used in this reaction with water–1ChCl/2Gly composite system as reaction medium (Table 1, entry 17). Unfortunately, ultrasonic acceleration has not been observed.
With the optimized reaction conditions in hand, the scope of the M–B–H reaction was investigated by employing a variety of aldehydes to react with acrylonitrile, methyl acrylate, ethyl acrylate or butyl acrylate. As shown in Table 2, both aromatic and aliphatic aldehydes can survive very efficient M–B–H reactions and give the corresponding M–B–H adducts in good to excellent yields. Among various aromatic aldehydes, molecules with electron-withdrawing groups (F, Cl and NO2) generally provide higher yields and shorter reaction time. It is worth mentioning that the aliphatic aldehydes also provide high yields under the present conditions (Table 2, entry 19). Interestingly, heteroaryl aldehydes and polycyclic aromatic aldehydes, also underwent M–B–H reaction to give the corresponding adducts with excellent yields (Table 2, entries 9–13). Another important observation that needs special mention is the reaction of benzene-1,4-dicarbaldehyde with acrylonitrile. In our previous work,22 we only got the M–B–H adduct of one aldehyde group, however, under the present conditions, when the reaction time is 40 min, we obtained a mixture of the M–B–H adduct of one aldehyde group and the M–B–H product of two aldehyde group, when the reaction time is 90 min, only the product of two aldehyde group reaction was obtained with 93% yield (Table 2, entry 18). When the methyl acrylate, ethyl acrylate and butyl acrylate were selected as activated alkenes (Table 2, entries 20–26), for the aromatic aldehydes with electron-withdrawing groups (Table 2, entries 21–23) and heteroaryl aldehydes (Table 2, entries 25–26), the good to excellent yields (82–88%) were obtained in short reaction time. Unfortunately, for the aromatic aldehydes with electron-donating groups, such as 3-hydroxy-4-methoxy benzaldehyde and 3-hydroxybenzaldehyde, the low yield 45% (Table 2, entry 14) and 38% was obtained (Table 2, entry 24). In addition, compared with acrylate, acrylonitrile shows higher reactivity. Thus the 1ChCl/2Gly DES–H2O composite system is indeed an alternative solvent media for M–B–H reaction.
|
||||
|---|---|---|---|---|
| Entry | R | EWG | Time (min) | Yieldb (%) |
| a All reactions were performed with aldehydes (10 mmol), activated alkenes (12 mmol) in the composite system (1 mL H2O and 3 g DES (1ChCl/2Gly)) in the presence of the catalyst DABCO (10 mmol) at room temperature. The reaction was monitored by TLC analysis.b Refers to isolated yield.c Acrylonitrile was 23 mmol. | ||||
| 1 | C6H5 | CN | 60 | 96 |
| 2 | 4-FC6H4 | CN | 90 | 93 |
| 3 | 4-ClC6H4 | CN | 80 | 98 |
| 4 | 2-ClC6H4 | CN | 80 | 92 |
| 5 | 4-NO2C6H4 | CN | 40 | 97 |
| 6 | 3-NO2C6H4 | CN | 50 | 95 |
| 7 | 2-CH3OC6H4 | CN | 120 | 86 |
| 8 | 4-CH3C6H4 | CN | 100 | 83 |
| 9 | 1-Naphthalyl | CN | 90 | 81 |
| 10 | 2-Pyridyl | CN | 60 | 94 |
| 11 | 2-Furanyl | CN | 80 | 79 |
| 12 | 4-Pyrazolyl | CN | 80 | 84 |
| 13 | 4-Quinolinyl | CN | 60 | 90 |
| 14 | 4-OH-3-CH3OC6H3 | CN | 210 | 45 |
| 15 | 4-Cl-3-NO2C6H3 | CN | 50 | 97 |
| 16 | 3,4-Cl2C6H3 | CN | 120 | 94 |
| 17 | 2,4-Cl2C6H3 | CN | 110 | 95 |
| 18c | 4-CHOC6H4 | CN | 90 | 93 |
| 19 | n-C3H7 | CN | 180 | 90 |
| 20 | C6H5 | COOCH3 | 120 | 78 |
| 21 | 4-ClC6H4 | COOCH3 | 80 | 83 |
| 22 | 4-NO2C6H4 | COOCH3 | 60 | 82 |
| 23 | 4-Cl-3-NO2C6H3 | COOCH3 | 80 | 88 |
| 24 | 3-OHC6H4 | COOCH3 | 180 | 38 |
| 25 | 4-Quinolinyl | COOCH2CH3 | 120 | 85 |
| 26 | 4-Quinolinyl | COOCH2CH2CH2CH3 | 120 | 86 |
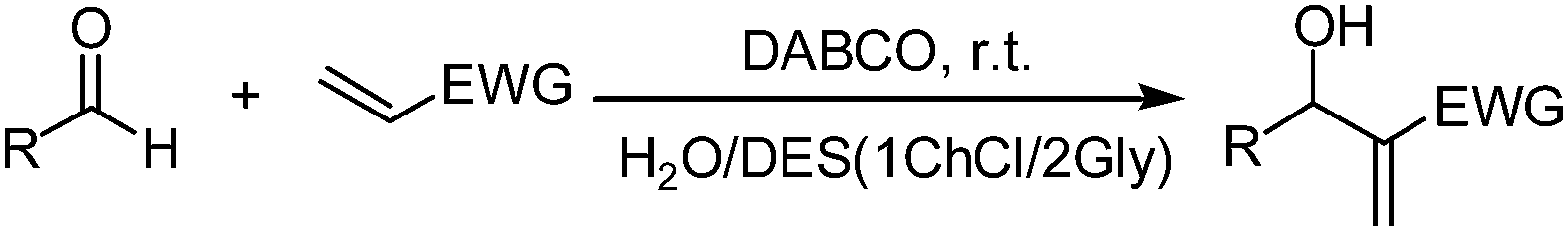
To evaluate the possibility of recycling the green solvent system 1ChCl/2Gly DES–H2O used for the M–B–H reaction, 4-chloro-benzaldehyde (10 mmol), acrylonitrile (12 mmol) and DABCO (10 mmol) were added to the composite system 1ChCl/2Gly DES–H2O (1 mL H2O and 3 g 1ChCl/2Gly DES). The reaction mixture was stirred at room temperature, and the reaction progress was monitored by thin layer chromatography (TLC) until aldehyde was consumed. At the end of reaction, the solid product suspended in the reaction mixture was filtered, washed with water and dried at room temperature under vacuum. Finally, the pure product was obtained. Then 4-chloro-benzaldehyde (10 mmol) and acrylonitrile (12 mmol) were added into the clear and colorless filtrate and the same reaction was repeated. Unfortunately, for the first cycle, 80 min later, only 62% yield was obtained and the 4-chloro-benzaldehyde was found in the reaction system. Why such a low yield was got? After careful observation to the crude product prepared by first run, we noticed that the crude product contains some water-soluble substance. To find out the truth, we carefully analyzed the 1H NMR spectra of the crude product and the purified product (see ESI†), the signal of the residual catalyst DABCO was found in the 1H NMR spectra of the crude product. The reason for the low yield may be that the loss of catalyst DABCO in the filtrate. In order to further clarify the reason, the DABCO catalyzed M–B–H reaction of 4-chloro-benzaldehyde with acrylonitrile in solvent system 1ChCl/2Gly DES–H2O was studied by IR spectral monitoring (for the IR spectra, please see ESI†). The spectra of the mixture of DES, H2O, DABCO, 4-chloro-benzaldehyde and acrylonitrile initially showed CN absorption at 2231 cm−1. But as the reaction progresses, the CN absorption peak becomes increasingly weaker. 80 minutes later, the CN absorption peak almost disappeared, which indicates that the reaction is complete, and at the same time, the present IR spectra of the reaction mixture is almost consistent with the IR spectra of the solvent system 1ChCl/2Gly DES–H2O, indicating that the solvent system 1ChCl/2Gly DES–H2O keeps its original structure very well and its nature has not changed. Since the reaction solvent is no problem, we add the catalyst DABCO (10 mmol) in the circular reaction. 80 min later, 97% yield was obtained again (Table 3, entry 1). To our delight, the recovered composite system was used at least six times almost without reduction of the reaction yields (Table 3, entries 1–6).
|
||
|---|---|---|
| Cycle | Time (min) | Yield (%) |
| a All reactions were performed with 4-chlorobenzaldehyde (10 mmol) and acrylonitrile (12 mmol) in the composite system (1 mL H2O and 3 g DES (1ChCl/2Gly)) in the presence of the catalyst DABCO (10 mmol) at room temperature. The reaction was monitored by TLC analysis. | ||
| 1 | 80 | 98 |
| 2 | 80 | 97 |
| 3 | 80 | 99 |
| 4 | 80 | 97 |
| 5 | 80 | 98 |
| 6 | 80 | 96 |
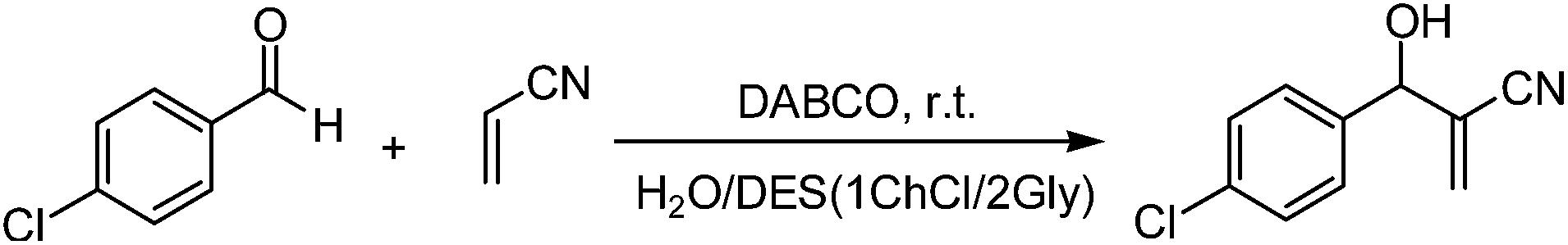
In the presence of the solvent system 1ChCl/2Gly DES–H2O, the mechanism for this reaction is complicated. However, based on our experimental results and literature reports,23 we reasoned that the solvent system 1ChCl/2Gly DES–H2O may has an increased efficiency for this reaction for two reasons. One is that the hydrogen-bond donors may activate the reaction by activation of carbonyl and enhancement the proton-transfer of intermediate, and the other is that the DES offers a polar ion environment for the stability of reaction intermediates. As in depicted in Scheme 1, first, the H-bond is formed between the hydrogen of hydroxyl in DES and the oxygen of carbonyl in aldehyde. The formed H-bonds make the eletrophilicity of carbonyl carbon stronger, which will benefits the attack of intermediate 1 and stable the intermediate 2. Then a ring transition state can be formed among the intermediate 2 and allow the proton-transfer to occur via a concerted step, in which DES–OH act as a shuttle to transfer the proton from the α-position to the alkoxide of int. 2.
 | ||
| Scheme 1 Possible the cyclic pathway for the DES (1ChCl/2Gly)–H2O–DABCO promoted M–B–H reaction. | ||
Conclusion
In summary, a recyclable DES solvent system, 1ChCl/2Gly DES–H2O, has been developed and used in the M–B–H reaction of a variety of aldehydes with activated alkenes. The composite solvent system (1ChCl/2Gly DES–H2O) could be readily prepared by simply mixing some cheap substance, such as glycerin, choline chloride and water. Under room temperature, the composite solvent system promoted M–B–H reactions proceeded very well, and the 1ChCl/2Gly DES–H2O solvent system could be recycled for at least 6 times showing no significant loss of activity. This protocol has notable advantages, such as low disposal costs, the ease of the work-up and reuse of the DES conveniently, which makes the present protocol practical for the preparation of multifunctional M–B–H products.Experimental
General
All reagents were obtained from commercial suppliers and used without further purification. The IR spectra were determined on an FTIR-8400 infrared spectrometer by dispersing samples in neat or KBr disks. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance 600 (600 MHz), 400 (400 MHz) spectrometer at ambient temperatures and using CDCl3 or DMSO-d6 as solvent. 1H and 13C NMR chemical shifts were reported in ppm relative to internal Me4Si. The elemental analyses were performed on a Vario EL Elemental Analyzer. Melting points were measured on WRS-1B digital melting point meter and are uncorrected.Preparation of DESs
According to the literature procedures,24 a series of DESs containing choline chloride were prepared by mixing choline chloride with urea, lactic acid or glycerol at molar ratios of choline chloride to urea, lactic acid or glycerol from 1:1 to 1:3 at 85–95 °C for 6–10 h until clear, transparent, homogeneous target liquids appeared.
General procedure for the M–B–H reaction
:5–1:3). The products were analyzed by NMR spectroscopy and the spectral data of all products are listed as follows.2-(Hydroxy-phenyl-methyl)-acrylonitrile (Table 2, entry 1)14d
Yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.44 (s, 5H), 6.13 (t, J = 6.0 Hz, 1H), 6.03 (s, 1H), 5.97 (s, 1H), 4.84 (s, 1H) ppm; 13C NMR (CDCl3, 101 MHz) δ 135.66, 130.76, 129.56, 129.20, 128.96, 127.66, 124.56, 78.14 ppm.2-[(4-Fluoro-phenyl)-hydroxy-methyl]-acrylonitrile (Table 2, entry 2)25
Yellow oil; 1H NMR (600 MHz, DMSO-d6) δ 7.44 (t, J = 8.9 Hz, 2H), 7.22 (t, J = 8.9 Hz, 2H), 6.35 (s, 1H), 6.21 (s, 1H), 6.12 (s, 1H), 5.35 (s, 1H) ppm; 13C NMR (151 MHz, DMSO-d6) δ 164.31, 140.82, 133.85, 131.68, 131.62, 130.41, 120.68, 118.56, 118.42, 75.41 ppm.2-[(4-Chloro-phenyl)-hydroxy-methyl]-acrylonitrile (Table 2, entry 3)
White crystal solid, mp 74.5 °C (lit.,19d 74.8–75.3 °C); 1H NMR (CDCl3, 400 MHz) δ 7.37 (d, J = 8.6 Hz, 2H), 7.25–7.33 (m, 2H), 6.08 (s, 1H), 6.02 (s, 1H), 5.25 (s, 1H), 3.12 (s, 1H) ppm; 13C NMR (CDCl3, 101 MHz) δ 137.68, 134.72, 130.40, 129.07, 127.92, 125.93, 116.77, 73.40 ppm.2-[(2-Chloro-phenyl)-hydroxy-methyl]-acrylonitrile (Table 2, entry 4)
White crystal solid, mp 101.2 °C (lit.,14d 100 °C); 1H NMR (CDCl3, 400 MHz) δ 7.64 (d, J = 7.6 Hz, 1H), 7.37 (dd, J = 12.9, 7.5 Hz, 2H), 7.30 (d, J = 10.0 Hz, 1H), 6.03 (s, 2H), 5.70 (s, 1H) ppm.2-[Hydroxy-(4-nitro-phenyl)-methyl]-acrylonitrile (Table 2, entry 5)
Yellow solid, mp 74.4 °C (lit.,14b 72–75 °C); 1H NMR (CDCl3, 400 MHz) δ 8.22 (d, J = 8.8 Hz, 2H), 7.60 (d, J = 8.5 Hz, 2H), 6.19 (s, 1H), 6.09 (s, 1H), 5.45 (s, 1H), 3.42 (s, 1H) ppm; 13C NMR (101 MHz, CDCl3) δ 147.95, 146.27, 131.30, 127.39, 125.49, 123.85, 116.47, 73.17 ppm.2-[Hydroxy-(3-nitro-phenyl)-methyl]-acrylonitrile (Table 2, entry 6)
Pale white solid, mp 65.2 °C (lit.,19d 64–66 °C); 1H NMR (600 MHz, DMSO-d6) δ 8.26 (s, 1H), 8.20 (d, J = 11.6 Hz, 1H), 7.85 (d, J = 7.7 Hz, 1H), 7.72 (t, J = 7.9 Hz, 1H), 6.66 (s, 1H), 6.32 (s, 1H), 6.21 (s, 1H), 5.55 (s, 1H) ppm; 13C NMR (151 MHz, CDCl3) δ 150.97, 146.76, 136.07, 135.17, 133.29, 129.83, 126.35, 124.18, 120.38, 75.24 ppm.2-[Hydroxy-(2-methoxy-phenyl)-methyl]-acrylonitrile (Table 2, entry 7)26
Yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.35 (dd, J = 18.9, 7.7 Hz, 2H), 7.01 (t, J = 7.5 Hz, 1H), 6.91 (d, J = 8.2 Hz, 1H), 5.99 (s, 1H), 5.96 (s, 1H), 5.51 (s, 1H), 3.83 (s, 3H), 3.58 (s, 1H) ppm; 13C NMR (101 MHz, CDCl3) δ 156.60, 129.89, 127.65, 127.32, 125.90, 121.09, 117.33, 110.96, 70.00, 55.44 ppm.2-[Hydroxy-(4-methyl-phenyl)-methyl]-acrylonitrile (Table 2, entry 8)27
Colorless oil; 1H NMR (400 MHz, CDCl3) δ 7.31 (d, J = 7.7 Hz, 2H), 7.23 (t, J = 7.7 Hz, 2H), 6.08 (d, J = 8.2 Hz, 1H), 6.02 (s, 1H), 5.25 (s, 1H), 3.41 (s, 3H), 2.40 (s, 3H) ppm.2-(Hydroxy-naphthalen-1-yl-methyl)-acrylonitrile (Table 2, entry 9)26
Yellow oil; 1H NMR (600 MHz, DMSO-d6) δ 8.19 (d, J = 9.2 Hz, 1H), 7.97 (d, J = 9.3 Hz, 1H), 7.92 (d, J = 8.2 Hz, 1H), 7.72 (d, J = 7.1 Hz, 1H), 7.59–7.51 (m, 3H), 6.46 (s, 1H), 6.35 (s, 1H), 6.20 (s, 1H), 6.04 (s, 1H) ppm; 13C NMR (151 MHz, DMSO-d6) δ 139.55, 136.79, 134.58, 133.39, 132.03, 131.96, 129.91, 129.56, 129.09, 128.74, 128.08, 127.01, 120.87, 73.31 ppm.2-(Hydroxy-pyridin-2-yl-methyl)-acrylonitrile (Table 2, entry 10)27
Yellow oil; 1H NMR (400 MHz, CDCl3) δ 8.61 (s, 1H), 7.80 (t, J = 7.5 Hz, 1H), 7.43 (d, J = 7.9 Hz, 1H), 7.34 (s, 1H), 6.26 (s, 1H), 6.09 (s, 1H), 5.33 (s, 1H), 1.74 (s, 1H) ppm; 13C NMR (101 MHz, CDCl3) δ 156.06, 148.51, 137.50, 130.99, 125.80, 123.74, 121.25, 116.76, 72.90 ppm.2-(Furan-2-yl-hydroxy-methyl)-acrylonitrile (Table 2, entry 11)27
Colorless oil; 1H NMR (400 MHz, CDCl3) δ 7.39 (d, J = 8.0 Hz, 1H), 6.44 (t, J = 7.9 Hz, 1H), 6.29 (d, J = 8.0 Hz, 1H), 6.19 (d, J = 4.0 Hz, 1H), 6.06 (s, 1H), 5.40 (s, 1H), 2.98 (br s, 1H) ppm.2-[Hydroxy-(1H-pyrazol-4-yl)-methyl]-acrylonitrile (Table 2, entry 12)
Colorless oil; 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 8.4 Hz, 2H), 7.65 (s, 1H), 6.47 (s, 2H), 5.64 (s, 1H), 4.47 (s, 1H) ppm. 13C NMR (101 MHz, CDCl3) δ 141.71, 133.45, 125.34, 124.46, 116.33, 113.67, 65.83. Anal. calcd for C7H7N3O: C, 56.37; H, 4.73; N, 28.17; found: C, 56.34; H, 4.75; N, 28.21.2-(Hydroxy-quinolin-4-yl-methyl)-acrylonitrile (Table 2, entry 13)
Yellow solid, mp 104.6 °C; 1H NMR (400 MHz, CDCl3) δ 8.68 (s, 1H), 8.03 (t, J = 8.7 Hz, 2H), 7.70–7.55 (m, 3H), 6.12 (s, 2H), 6.06 (s, 1H) ppm; 13C NMR (CDCl3, 101 MHz) δ 149.84, 147.67, 145.25, 131.83, 129.82, 129.56, 127.33, 125.43, 125.25, 123.29, 119.24, 116.88, 70.03 ppm. Anal. calcd for C13H10N2O: C, 74.27; H, 4.79; N, 13.33; found: C, 74.31; H, 4.77; N, 13.38.2-[Hydroxy-(4-hydroxy-3-methoxy-phenyl)-methyl]-acrylonitrile (Table 2, entry 14)19a
Pale yellow solid, mp 110.6 °C; 1H NMR (400 MHz, CDCl3) δ 7.32 (s, 1H), 7.29 (s, 1H), 7.18 (s, 1H), 5.08 (s, 2H), 4.16 (s, 1H), 3.97 (s, 3H) ppm; 13C NMR (101 MHz, CDCl3) δ 148.35, 141.74, 134.90, 129.35, 124.42, 122.20, 119.15, 118.05, 74.72, 55.96. Anal. calcd for C11H11NO3: C, 64.38; H, 5.40; N, 6.83; found: C, 64.34; H, 5.42; N, 6.79.2-[Hydroxy-(4-chloro-3-nitro-phenyl)-methyl]-acrylonitrile (Table 2, entry 15)14d
Yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.76 (s, 2H), 7.39 (s, 2H), 6.04 (s, 1H), 5.90 (s, 1H), 5.21 (s, 1H), 3.00 (s, 1H) ppm; 13C NMR (101 MHz, CDCl3) δ 150.99, 142.85, 134.72, 133.93, 130.58, 129.07, 127.92, 126.00, 116.85, 74.18 ppm.2-[(3,4-Dichloro-phenyl)-hydroxy-methyl]-acrylonitrile (Table 2, entry 16)25
Yellow oil; 1H NMR (600 MHz, DMSO-d6) δ 7.67 (d, J = 8.3 Hz, 1H), 7.63 (d, J = 2.0 Hz, 1H), 7.38 (dd, J = 8.6, 2.3 Hz, 1H), 6.53 (s, 1H), 6.26 (d, J = 1.0 Hz, 1H), 6.17 (s, 1H), 5.39 (d, J = 4.2 Hz, 1H) ppm; 13C NMR (151 MHz, DMSO-d6) δ 145.60, 134.92, 134.47, 134.07, 133.85, 131.52, 129.89, 129.54, 120.42, 74.75 ppm.2-[(2,4-Dichloro-phenyl)-hydroxy-methyl]-acrylonitrile (Table 2, entry 17)
White solid, mp 73.5 °C (lit.,14d 74 °C); 1H NMR (400 MHz, CDCl3) δ 7.55 (d, J = 8.01, 1H), 7.38 (s, 1H), 7.31 (d, J = 8.01 Hz, 1H), 6.08 (s, 1H), 5.69 (s, 1H), 3.12 (s, 1H) ppm; 13C NMR (101 MHz, CDCl3) δ 141.67, 137.14, 136.07, 134.81, 130.29, 129.22, 128.12, 125.76, 118.42, 72.20 ppm.2-{[4-(2-Cyano-1-hydroxy-allyl)-phenyl]-hydroxy-methyl}-acrylonitrile (Table 2, entry 18)
White crystal solid, mp 134.2 °C (lit.,19d 134.8–135.3 °C); 1H NMR (400 MHz, CDCl3) δ 7.48 (s, 5H), 6.17 (s, 2H), 6.08 (s, 2H), 5.37 (s, 2H), 3.77 (s, 2H) ppm; 13C NMR (101 MHz, DMSO-d6) δ 141.17, 130.96, 127.46, 126.76, 117.87, 72.86 ppm.3-Hydroxy-2-methylene-hexanenitrile (Table 2, entry 19)19a
Yellow oil; 1H NMR (600 MHz, DMSO-d6) δ 6.03 (d, J = 6.3 Hz, 2H), 5.50 (s, 1H), 4.10 (s, 1H), 1.48 (dd, J = 15.1, 7.4 Hz, 2H), 1.35–1.25 (m, 2H), 0.87 (d, J = 7.4 Hz, 3H) ppm; 13C NMR (151 MHz, DMSO-d6) δ 133.62, 130.80, 121.05, 73.86, 43.37, 43.23, 43.09, 42.95, 42.82, 42.68, 42.54, 40.82, 21.30, 17.03 ppm.2-(Hydroxy-phenyl-methyl)-acrylic acid methyl ester (Table 2, entry 20)14b
Colorless oil; 1H NMR (400 MHz, CDCl3) δ 7.32–7.42 (m, 5H), 6.28 (s, 1H), 5.86 (s, 1H), 5.45 (s, 1H), 3.67 (s, 3H), 3.10 (s, 1H) ppm.2-[(4-Chloro-phenyl)-hydroxy-methyl]-acrylic acid methyl ester (Table 2, entry 21)
White solid; mp 44.1 °C (lit.,14b 43–44 °C); 1H NMR (400 MHz, CDCl3) δ 7.30 (s, 4H), 6.33 (s, 1H), 5.86 (s, 1H), 5.50 (s, 1H), 3.72 (s, 3H), 3.32 (s, 1H) ppm.2-[Hydroxy-(4-nitro-phenyl)-methyl]-acrylic acid methyl ester (Table 2, entry 22)
Yellow solid, mp 72.3 °C (lit.,14b 71–73 °C); 1H NMR (400 MHz, CDCl3) δ 8.19 (d, J = 8.8 Hz, 2H), 7.57 (d, J = 8.7 Hz, 2H), 6.40 (s, 1H), 5.90 (s, 1H), 5.63 (s, 1H), 3.74 (s, 3H), 3.51 (s, 1H) ppm; 13C NMR (101 MHz, CDCl3) δ 166.39, 148.69, 147.40, 140.98, 127.39, 127.26, 123.62, 72.60, 52.24 ppm.2-[(4-Chloro-3-nitro-phenyl)-hydroxy-methyl]-acrylic acid methyl ester (Table 2, entry 23)
White solid, mp 85.4 °C (lit.,14d 84 °C); 1H NMR (400 MHz, CDCl3) δ 7.92 (s, 1H), 7.55 (q, J = 8.4 Hz, 2H), 6.42 (s, 1H), 5.94 (s, 1H), 5.59 (s, 1H), 3.76 (s, 3H), 3.45 (s, 1H) ppm; 13C NMR (101 MHz, CDCl3) δ 166.25, 147.87, 142.17, 140.70, 131.76, 131.16, 127.36, 126.11, 123.61, 72.05, 52.25 ppm.2-[Hydroxy-(3-hydroxy-phenyl)-methyl]-acrylic acid methyl ester (Table 2, entry 24)19a
Pale yellow solid, mp 100.4 °C; 1H NMR (400 MHz, CDCl3) δ 7.22 (t, J = 7.8 Hz, 1H), 6.91 (d, J = 12.3 Hz, 2H), 6.78 (s, 1H), 6.36 (s, 1H), 5.86 (s, 1H), 5.66 (s, 1H), 5.53 (s, 1H), 3.75 (s, 3H), 3.27 (s, 1H) ppm; 13C NMR (101 MHz, CDCl3) δ 166.87, 155.81, 143.16, 141.73, 129.75, 126.54, 118.87, 114.87, 113.45, 73.13, 51.89 ppm. Anal. calcd for C11H12O4: C, 63.45; H, 5.81; found: C, 63.34; H, 5.75.2-(Hydroxy-quinolin-4-yl-methyl)-acrylic acid ethyl ester (Table 2, entry 25)
White solid, mp 87.4 °C; 1H NMR (400 MHz, CDCl3) δ 8.80 (s, 1H), 8.06 (s, 1H), 7.97 (s, 1H), 7.67–7.51 (m, 2H), 6.38 (s, 2H), 5.64 (s, 1H), 4.21 (s, 1H), 2.60 (s, 1H), 1.24–1.12 (m, 3H) ppm; 13C NMR (101 MHz, CDCl3) δ 166.34, 150.11, 147.98, 147.40, 141.81, 129.82, 129.19, 127.28, 126.68, 125.93, 123.83, 118.98, 67.74, 61.22, 14.06 ppm. Anal. calcd for C15H15NO3: C, 70.02; H, 5.88; N, 5.44; found: C, 70.26; H, 5.91; N, 5.48 ppm.2-(Hydroxy-quinolin-4-yl-methyl)-acrylic acid butyl ester (Table 2, entry 26)
White solid, mp 104.2 °C; 1H NMR (400 MHz, CDCl3) δ 8.88 (d, J = 4.5 Hz, 1H), 8.12 (d, J = 8.4 Hz, 1H), 7.96 (d, J = 8.4 Hz, 1H), 7.70 (t, J = 7.7 Hz, 1H), 7.61 (d, J = 4.5 Hz, 1H), 7.54 (t, J = 7.6 Hz, 1H), 6.39 (s, 2H), 5.57 (s, 1H), 4.20 (t, J = 5.1 Hz, 2H), 1.66–1.59 (m, 2H), 1.36–1.29 (m, 2H), 0.91 (t, J = 7.4 Hz, 3H) ppm; 13C NMR (101 MHz, CDCl3) δ 166.54, 150.21, 148.03, 146.55, 141.27, 130.00, 129.25, 127.72, 126.78, 125.85, 123.72, 118.94, 68.32, 65.26, 30.49, 19.12, 13.66 ppm. Anal. calcd for C17H19NO3: C, 71.56; H, 6.71; N, 4.91; found: C, 71.48; H, 6.67; N, 4.95 ppm.Reusability and recovery of the composite solvent system 1ChCl/2Gly DES–H2O
After the first run of the reaction was completed, the product is obtained by filtering or the product was extracted by diethyl ether into the organic layer, and the remained composite solvent system 1ChCl/2Gly DES–H2O was directly reused for the next cycle of the reaction.Acknowledgements
We gratefully acknowledge financial support from the Fund for Shanxi Key Subjects Construction (No. 20141010), the Key Subjects Construction of Xinzhou Teachers University (No. XK201304) and the Natural Science Foundation of Shanxi Province (No. 2016021007-3).Notes and references
- S. E. Drewes and G. H. P. Roos, Tetrahedron, 1988, 44, 4653–4670 CrossRef CAS.
- V. K. Aggarwal and A. Mereu, Chem. Commun., 1999, 35, 2311–2312 RSC.
- V. K. Aggarwal, I. Emme and S. Y. Fulford, J. Org. Chem., 2003, 68, 692–700 CrossRef CAS PubMed.
- (a) D. Basavaiah, P. D. Rao and R. S. Hyma, Tetrahedron, 1996, 52, 8001–8062 CrossRef CAS; (b) D. Basavaiah, J. Rao and T. Satyanarayana, Chem. Rev., 2003, 103, 811–892 CrossRef CAS PubMed.
- D. Basavaiah, B. S. Reddy and S. S. Badsara, Chem. Rev., 2010, 110, 5447–5674 CrossRef CAS PubMed.
- B. Pawar, V. Padalkar, K. Phatangare, S. Nirmalkara and A. Chaskar, Catal. Sci. Technol., 2011, 1, 1641–1644 Search PubMed.
- B. A. Shairgojray, A. A. Dar and B. A. Bhat, Tetrahedron Lett., 2013, 54, 2391–2394 CrossRef CAS.
- (a) A. Fall, I. Seck, O. Diouf, M. Gaye, M. Seck, G. Gómez and Y. Fall, Tetrahedron Lett., 2015, 56, 5128–5131 CrossRef CAS; (b) D. Mendoza-Espinosa, R. González-Olvera, C. Osornio, G. E. Negrón-Silva and R. Santillan, New J. Chem., 2015, 39, 1587–1591 RSC; (c) Y. Jeong and J. S. Ryu, J. Org. Chem., 2010, 75(12), 4183–4191 CrossRef CAS PubMed; (d) J. C. Hsu, Y. H. Yen and Y. H. Chu, Tetrahedron Lett., 2004, 45, 4673–4676 CrossRef CAS; (e) A. Kumar and S. S. Pawar, J. Mol. Catal. A: Chem., 2004, 211, 43–47 CrossRef CAS; (f) J. N. Rosa, C. A. M. Afonso and A. G. Santos, Tetrahedron, 2001, 57, 4189–4193 CrossRef CAS.
- S. Chandrasekhar, C. Narsihmulu, B. Saritha and S. Shameem Sultana, Tetrahedron Lett., 2004, 45, 5865–5867 CrossRef CAS.
- P. R. Krishna, A. Manjuvani, V. Kannan and G. V. M. Sharma, Tetrahedron Lett., 2004, 45, 1183–1185 CrossRef CAS.
- P. M. Rose, A. A. Clifford and C. M. Rayner, Chem. Commun., 2002, 38, 968–969 RSC.
- J. N. Tan, M. Ahmar and Y. Queneau, RSC Adv., 2015, 5, 69238–69242 RSC.
- P. N. Joshi, L. Purushottam, N. K. Das, S. Mukherjee and V. Rai, RSC Adv., 2016, 6, 208–211 RSC.
- (a) C. Yu, B. Liu and L. Hu, J. Org. Chem., 2001, 66(16), 5413–5418 CrossRef CAS PubMed; (b) J. Cai, Z. Zhou, G. Zhao and C. Tang, Org. Lett., 2002, 4, 4723–4725 CrossRef CAS PubMed; (c) V. K. Aggarwal, D. K. Dean, A. Mereu and R. Williams, J. Org. Chem., 2002, 67(2), 510–514 CrossRef CAS PubMed; (d) S.-H. Zhao, H.-Y. Bie and Z.-B. Chen, Org. Prep. Proced. Int., 2005, 37, 231–237 CrossRef CAS; (e) R. O. M. A. de Souza, V. L. P. Pereira, P. M. Esteves and M. L. A. A. Vasconcellos, Tetrahedron Lett., 2008, 49, 5902–5905 CrossRef CAS; (f) G. L. Li, K. K. Y. Kung, L. Zou, H. C. Chong, Y. C. Leung, K. H. Wong and M. K. Wong, Chem. Commun., 2012, 48, 3527–3529 RSC; (g) F. Yi, X. Zhang, H. Sun and S. Chen, Acta Chim. Sin., 2012, 70(6), 741–746 CrossRef CAS.
- J. Mack and M. Shumba, Green Chem., 2007, 9, 328–330 RSC.
- Q. Zhang, K. D. Vigier, S. Royer and F. Jérôme, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- (a) P. Abbott, E. I. Ahmed, R. C. Harris and K. S. Ryder, Green Chem., 2014, 16, 4156–4161 RSC; (b) D. V. Wagle, H. Zhao and G. A. Baker, Acc. Chem. Res., 2014, 47, 2299–2308 CrossRef CAS PubMed.
- (a) C. Russ and B. Koenig, Green Chem., 2012, 14, 2969–2982 RSC; (b) M. Fransisco, A. van den Bruinhorst and M. C. Kroon, Angew. Chem., Int. Ed., 2013, 52, 3074–3085 CrossRef PubMed; (c) Y. Dai, J. van Spronsen, G. J. Witkamp, R. Verpoorte and Y. H. Choi, Anal. Chim. Acta, 2013, 766, 61–68 CrossRef CAS PubMed; (d) P. Liu, J. W. Hao, L. P. Mo and Z. H. Zhang, RSC Adv., 2015, 5, 48675–48704 RSC; (e) H. C. Hu, Y. H. Liu, B. L. Li, Z. S. Cui and Z. H. Zhang, RSC Adv., 2015, 5, 7720–7728 RSC.
- (a) S. H. Zhao, H. R. Zhang, L. H. Feng and Z. B. Chen, J. Mol. Catal. A: Chem., 2006, 258, 251–256 CrossRef CAS; (b) S. Zhao, E. Zhao, P. Shen, M. Zhao and J. Sun, Ultrason. Sonochem., 2008, 15, 955–959 CrossRef CAS PubMed; (c) S. Zhao, X. Wang and L. Zhang, RSC Adv., 2013, 3, 11691–11696 RSC; (d) S. Zhao, D. Wang, M. Wang, J. Kang and L. Zhang, Chin. J. Org. Chem., 2015, 35, 865–874 CrossRef CAS.
- (a) C. J. Li and L. Chen, Chem. Soc. Rev., 2006, 35, 68–82 RSC; (b) M. O. Simon and C. J. Li, Chem. Soc. Rev., 2012, 41, 1415–1427 RSC.
- A. Gaplovsky, M. Gaplovsky, S. Toma and J. L. Luche, J. Org. Chem., 2000, 65, 8444–8447 CrossRef CAS PubMed.
- S. H. Zhao, Q. J. Zhang, X. E. Duan and L. H. Feng, Synth. Commun., 2011, 41, 3289–3297 CrossRef CAS.
- (a) R. Robiette, V. K. Aggarwal and J. N. Harvey, J. Am. Chem. Soc., 2007, 129, 15513–15525 CrossRef CAS PubMed; (b) S. Zhao, M. He, Z. Guo, N. Zhou, D. Wang, J. Li and L. Zhang, RSC Adv., 2015, 5, 32839–32845 RSC; (c) A. Singh and A. Kumar, J. Org. Chem., 2012, 77(19), 8775–8779 CrossRef CAS PubMed.
- (a) Q. Wang, X. Yao, Y. Geng, Q. Zhou, X. Lu and S. Zhang, Green Chem., 2015, 17, 2473–2479 RSC; (b) J. Li, Z. Han, Y. Zou and B. Yu, RSC Adv., 2015, 5, 93937–93944 RSC.
- R. O. M. A. de Souza, B. A. Meireles, L. C. S. Aguiar and M. L. A. A. Vasconcellos, Synthesis, 2004, 10, 1595–1600 Search PubMed.
- H. Gong, C. Q. Cai, N. F. Yang, L. W. Yang, J. Zhang and Q. H. Fan, J. Mol. Catal. A: Chem., 2006, 249, 236–239 CrossRef CAS.
- X. Mi, S. Luo, H. Xu, L. Zhang and J. P. Cheng, Tetrahedron, 2006, 62, 2537–2544 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of the M–B–H products. See DOI: 10.1039/c6ra04710f |
| This journal is © The Royal Society of Chemistry 2016 |