
Membranes combining chitosan and natural-origin nanoliposomes for tissue engineering†
Abstract
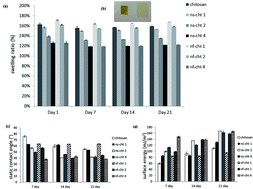
Chitosan thin films, elaborated by solvent casting, were functionalized by incorporating nanoliposomes based on natural vegetable (soy based) and marine (salmon derived) lecithin. The marine lecithin used in this study contains a higher percentage of polyunsaturated fatty acids (PUFAs) and polar lipids compared with vegetal lecithin. The physical-chemistry properties of the obtained films were characterized by water contact angle (WCA), Fourier Transform InfraRed spectroscopy (FT-IR), water uptake test, and Torsional Harmonic Atomic Force Microscopy analysis (TH-AFM). The surface wettability, swelling ratio, roughness and local stiffness of the thin films can be modified and controlled by adding nanoliposomes. The WCA decreased with the increase of the amount of nanoliposomes. Equilibrium water uptakes of about 170% were achieved in 24 h for the different formulations. The FT-IR results showed the existence of chemical interactions between chitosan and nanoliposomes. The surface topography of the films were identical in terms of asymmetry and amplitude distribution of roughness measurements but showed a significant increase of asperity height when incorporating soya nanoliposomes. This variation is accompanied with a decrease in the average of surface rigidity and of adhesive force value, resulting in a heterogeneous surface. The behaviour of human mesenchymal stem cells (hMSCs) cultured on the films was investigated. Results showed that the films favor cell proliferation when the concentration of soya and salmon nanoliposomes is below 2 mg mL−1 and 4 mg mL−1, respectively. The highest cell proliferation of hMSCs culture was observed when the concentration of salmon nanoliposomes was 1 mg mL−1. This work provides evidence that nanoliposome-functionalized chitosan thin films could offer adequate cyto-friendly cell culture supports for hMSCs, and may potentially be used as suitable scaffolds for tissue engineering applications.
 Please wait while we load your content...
Please wait while we load your content...

