
Supercritical fluid assisted biotemplating synthesis of Si–O–C microspheres from microalgae for advanced Li-ion batteries†
Yang Xiaa,
Ruyi Fanga,
Zhen Xiaob,
Luoyuan Ruana,
Rongjun Yanc,
Hui Huanga,
Chu Lianga,
Yongping Gana,
Jun Zhanga,
Xinyong Tao*a and
Wenkui Zhang*a
aCollege of Materials Science and Engineering, Zhejiang University of Technology, Hangzhou, 310014, China. E-mail: tao@zjut.edu.cn; msechem@zjut.edu.cn
bCollege of Materials Science and Engineering, China Jiliang University, Hangzhou, 310018, China
cOcean College, Zhejiang University of Technology, Hangzhou, 310014, China
First published on 18th July 2016
Abstract
Silicon oxycarbide (Si–O–C) materials with high specific capacity are considered as a promising anodic material alternative to commercial graphite for advanced Li-ion batteries. However, the rapid capacity fading and poor rate performance are the main obstacles for practical application and still remain a large challenge. In this work, microalgae (Nannochloropsis) served as a biological template and carbon source to synthesize Si–O–C microspheres with the assistance of supercritical CO2 fluid. Compared to conventional artificial templates, microalgae is abundant, renewable and available, and can be regarded as a promising biological template. Meanwhile, supercritical CO2 fluid with high penetration, high diffusivity and high dissolving capacity can serve as a superior solvent to guarantee the efficient mass transfer and uniform dispersion of precursors. As anodic materials for Li-ion batteries, Si–O–C microspheres exhibit a high reversible specific capacity of 450 mA h g−1 at a current density of 0.1 A g−1 over 200 cycles, excellent rate cycling stability and high coulombic efficiency (100%). The discovery of this novel strategy to fabricate Si–O–C materials presents possibilities for energy storage applications.
1. Introduction
The rapidly rising market demands of portable electronic devices, electric vehicles and large-scale energy storage have spurred tremendous efforts on the development of advanced rechargeable Li-ion batteries.1–3 However, graphitic carbon as the commercial anode material often suffers from several intractable shortcomings, especially low specific capacity, poor cycling stability and inferior rate capability, which strongly restrict its widespread application.4 In order to overcome the above issues, great efforts have been focused on exploring new anodic materials to replace graphitic carbon, which is the crucial step toward the realization of high-performance Li-ion batteries.In this respect, silicon oxycarbides (Si–O–C) have a complex amorphous structure with disorderly mixing silicon oxycarbide units (SiCnO4−n, 0 ≤ n ≤ 4, i.e. SiO4, SiCO3, SiC2O2, SiC3O and SiC4) and free carbon network,5,6 which have received intensive attention as an alternative anodic material to commercial graphitic carbon. The pioneering work of Dahn research group7,8 demonstrated that the Li storage capacity of Si–O–C material is closely related to the phase composition and microstructure. According to previous works,5,9–11 the reversible Li storage sites of Si–O–C materials mainly arise from the edges and interstitial spaces of graphene layers, directly or indirectly amorphous Si–O–C phases, micro-/nano-pores and interfacial and defect sites. Obviously, Si–O–C materials can store more Li compared to the conventional graphitic carbon materials since the maximum Li storage capacity of graphitic carbon is only 372 mA h g−1 (Cgraphite + xLi+ + xe− ↔ LixC6, xmax = 1). Therefore, the successful implementation of controllable tailoring the phase composition and microstructure of Si–O–C materials might be the key factor to achieve high electrochemical performance. So far, a variety of methods to produce Si–O–C materials have been reported, such as pyrolysis,12–15 sol-gels,16,17 and electrodeposition.18,19 For instance, Kaspar et al.12 reported that nano-crystalline and nano-amorphous silicon particles were successfully embedded within carbon-rich silicon oxycarbide matrix by a pyrolysis process, presenting a stable cycling performance up to 100 cycles. Meanwhile, Pradeep et al.14 synthesized carbon-rich Si–O–C ceramics derived from polysiloxane via a pyrolysis process at 1000 °C. Furthermore, Liu et al.16 prepared high capacity Si–O–C materials using a sol–gel method, which exhibited a stable reversible capacity of 900 mA h g−1 at 50 mA g−1. Osaka et al.18,19 developed a novel Si–O–C composite thick film anodic material by electrodeposition from an organic solvent with a complex structure current collector, which substantially improved electrochemical performance both in specific capacity and cycle ability.
Different from the above methods, biotemplating technique is considered as a high-efficiency strategy for fabricating various functional materials with uniform size, multiform morphology, and elaborate microstructure. Inspired by the abundant, inexpensive, and renewable nature biomaterials, our group has devoted to the utilization of various biomaterials including bamboos,20–23 cotton fibers,24–26 kapok fibers,27,28 pollens,29,30 microalgaes31–33 as both biologic templates and carbon sources to synthesize carbon-based electrode materials, which could circumvent the fundamental issues of electrode materials in energy storage and conversion fields and achieve high electrochemical performance. Particularly, microalgaes possess many merits (i.e. easy availability, low cost, uniform and multiform geometries) compared to other artificial/biological templates, which are ideal candidates for the design and fabrication of novel electrode materials with controllable morphology and microstructure.31–33
In this work, a new synthetic strategy combined biotemplating method and supercritical CO2 fluid technology is proposed to prepare Si–O–C materials. Since supercritical CO2 fluid exhibits both “gas-like” and “liquid-like” unique features, it possesses low viscosity, high diffusivity and excellent dissolving capacity.34 Taking these specific properties, supercritical CO2 fluid is expected to be superior solvent to penetrate into target materials and control mass transfer with a high efficiency. Hence, we select Nannochloropsis microalages as both biologic templates and carbon sources to synthesize high performance Si–O–C microspheres with the assistance of supercritical CO2 fluid. Generally, Nannochloropsis cells are primarily composed of moisture, available carbohydrates, crude proteins, lipids, ash, fiber, nucleic acids, and pigments (carotenoids and carbohydrates).31,33 On average, the carbohydrates, protein and lipids are the main components, which are 37.6%, 28.8% and 18.4% based on dry biomass, respectively.35 Therefore, Nannochloropsis microalages will be a natural carbon source for synthesizing Si–O–C microspheres. The main process of the synthesis of Si–O–C microspheres is described in Fig. 1. It is believed that this novel strategy will stimulate more investigations of Si–O–C materials, and also extend the scope of both biotemplating method and supercritical CO2 fluid technology to other functional materials.
 | ||
| Fig. 1 Schematic illustration of the preparation process of Si–O–C microspheres based on biotemplating method with the assistance of supercritical CO2 fluid. | ||
2. Experimental section
2.1 Microalgae culture and pre-treatment
In this work, Nannochloropsis was purchased from Marine Biological Culture Collection Centre (China). The seed culture medium and the culture conditions were the same as those used in our previous work.31 After harvesting, the obtained microalgaes washed several times in deionized water to remove the unwanted materials. In order to replicate the morphology and microstructure, microalgaes were added into a 50 mL mixed solution of alcohol and formaldehyde (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in volume), and violently stirred for 30 min. Finally, the resulting microalgaes were filtered and washed with deionized water and dried at 80 °C for 4 h in air.
:1 in volume), and violently stirred for 30 min. Finally, the resulting microalgaes were filtered and washed with deionized water and dried at 80 °C for 4 h in air.
2.2 Synthesis of Si–O–C microspheres and microalgae carbon
In a typical procedure, 1.5 g pretreated microalgaes and 4.4 mL tetraethyl orthosilicate (TEOS) (28.4%) were added into a 100 mL stainless steel milling jar. Subsequently, CO2 was pumped into the milling jar until the pressure reached 8.5 MPa. Then, the milling jar was milled on a planetary ball mill (Nanjing, QM-1SP2) at 350 rpm. During the milling process, the ambient temperature should be strictly kept at 32 °C. After milling for 12 h, the gas was released immediately. Finally, the obtained precursor was placed inside an alumina boat and calcined in a tube furnace at 500 °C for 4 h under a flowing argon atmosphere. For comparison, microalgae carbon was calcined without adding TEOS and using supercritical CO2 fluid at the same conditions to obtain the control sample.2.3 Materials characterizations
X-ray diffraction (XRD) patterns were carried out by a Rigaku Ultima IV Powder X-ray diffractometer using Cu Kα radiation (λ = 0.15418 nm). Fourier transform infrared radiation (FT-IR) analysis was conducted on an infrared spectrophotometer (Nicolet 6700) by the KBr pellet method in air. Raman spectra were recorded by a Renishaw InVia Raman spectroscopy (λ = 532 nm). X-ray photoelectron spectroscopy (XPS) measurements were performed on an Axis Ultra DLD system (Kratos) with a monochromatic Al-Kα (1486.6 eV) X-ray source. The morphology and microstructure were observed by scanning electron microscopy (SEM, Hitachi S-4800), transmission electron microscopy (TEM, FEI, Tecnai G2 F30) with an energy dispersive spectroscopy (EDS) detector.2.4 Electrochemical measurements
The electrochemical performance of samples was investigated using CR2025 coin-type cells assembled in an argon-filled glove box. The electrodes were comprised of 80 wt% samples, 10 wt% Super-P and 10 wt% carboxymethylcellulose (CMC) dissolved in water at 80 °C as binder. The resultant viscous slurry was coated on copper foil and dried at 120 °C under vacuum for 12 h. The lithium foil was used as the counter electrode, 1 M solution of LiPF6 in ethylene carbonate and dimethyl carbonate (1:1 in volume) was used as the electrolyte with Celgard 2300 as the separator. Cyclic voltammograms (CV) were recorded on a CHI650B electrochemical workstation (Chenhua, Shanghai, China) with a voltage range from 0 to 3.0 V at a scan rate of 0.1 mV s−1. The galvanostatic charge–discharge tests were conducted on a Neware battery test system (Shenzhen Neware Technology Co. Ltd.) in the voltage range of 0.01–3.0 V at room temperature. The current densities and specific capacities were calculated based on the mass of Si–O–C composite microspheres. Electrochemical impedance spectroscopy (EIS) were carried out in the frequency range from 0.1 to 106 Hz with a 5 mV amplitude at the open-circuit voltage of fresh cells by a Ivium-n-Stat workstation.
3. Results and discussion
Fig. 2a shows XRD patterns of microalgae carbon and Si–O–C microspheres. Only one broad peak can be detected around 2θ = 26°, implying microalgae carbon and Si–O–C microspheres have the amorphous structure after the calcination. This result also indicates that there are no crystalline Si or SiOx particles in Si–O–C microspheres. To better understand the chemical bonds, FT-IR analysis was carried out. As shown in Fig. 2b, the spectra of microalgae carbon and Si–O–C microspheres both exhibit the absorption bands at v = 1610 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), v = 1570, 1360, 1070 cm−1 (C–O), v = 1090, 1020, 950 cm−1 (Si–O–C), v = 1120, 770 cm−1 (Si–O–Si), respectively.5,36,37 Interestingly, Si–O–C and Si–O–Si groups also exist in microalgae carbon, however the intensity is much weaker than that of Si–O–C microspheres. Since Si is a very important element for microalgaes, therefore the trace Si element will serve as a natural Si source for the formation of Si–O–C and Si–O–Si bonds during the calcination.
C), v = 1570, 1360, 1070 cm−1 (C–O), v = 1090, 1020, 950 cm−1 (Si–O–C), v = 1120, 770 cm−1 (Si–O–Si), respectively.5,36,37 Interestingly, Si–O–C and Si–O–Si groups also exist in microalgae carbon, however the intensity is much weaker than that of Si–O–C microspheres. Since Si is a very important element for microalgaes, therefore the trace Si element will serve as a natural Si source for the formation of Si–O–C and Si–O–Si bonds during the calcination.
 | ||
| Fig. 2 (a) FTIR spectra of microalgae carbon and Si–O–C microspheres. (b) XRD patterns of microalgae carbon and Si–O–C microspheres. | ||
Raman spectroscopy is a useful tool for characterizing carbon based materials, particularly for estimating the quality of carbon. Fig. 3a shows Raman spectra of microalgae carbon and Si–O–C microspheres. Si–Si peaks located at 500 cm−1 (first Si–Si vibrational mode) and 930 cm−1 (second Si–Si vibrational mode) are not obtained in microalgae carbon nor Si–O–C microspheres.38 This finding is matching well with XRD and FTIR results as shown in Fig. 2. Two significant peaks around at 1350 and 1580 cm−1 can be noticed both in microalgae carbon and Si–O–C microspheres, implying the existent of the disordered carbon (D-band) and graphite like sp2 carbon (G-band). Generally, the ratio of ID and IG can be used to evaluate the disordering degree of carbonaceous materials, which also is associated with the electronic conductivity. After a careful comparison, the ID/IG ratio of microalgae carbon (ID/IG = 0.87) is smaller than that of Si–O–C microspheres (ID/IG = 0.99), indicating that the addition of Si element into microalgae carbon will increase the disordering degree.
 | ||
| Fig. 3 (a) Raman spectra of microalgae carbon and Si–O–C microspheres. (b) XPS survey spectra of microalgae carbon and Si–O–C microspheres. (c) and (d) High-resolution C 1s and Si 2p XPS spectra of Si–O–C microspheres. | ||
In order to obtain the detailed information about the elemental character and chemical state of microalgae carbon and Si–O–C microspheres, X-ray photoelectron spectroscopy (XPS) measurements are performed. In this work, the C 1s peak (284.8 eV) is used to do the calibration. Fig. 3b is the comparison of the XPS survey spectra between microalgae carbon and Si–O–C microspheres. The characteristic peaks are C, O, Si and N. The trace N element detected in both microalgae carbon and Si–O–C microspheres may result from microalgaes since N element is related to many life activities.33 However, the content of N element is extremely low, which can almost be ignored. Moreover, the intensity of Si and O in Si–O–C microspheres is much stronger than that in microalgae carbon. The concentration (in at%) of Si, O and C in Si–O–C microspheres is 10.61%, 31.51% and 57.88%, respectively. As indicated in Fig. 3c, the C 1s spectrum of microalgae carbon could be curve-fitted into two peaks with bonding energies of 286.5 eV (C–O bond) and 284.8 eV (C–C/CC bond). However, the C 1s spectrum of Si–O–C microspheres clearly demonstrates three peaks at 284.8, 285.3 and 286.5 eV, which can be ascribed to C–C/CC, C–O–Si and C–O bonds, respectively.39 Since the Si 2p signal of microalgae carbon is very weak as shown in the XPS survey spectrum (Fig. 3b). Herein, we only present the Si 2p spectrum of Si–O–C microspheres (Fig. 3d). In the Si 2p spectrum of Si–O–C microspheres, the fitting peak at 103.9 eV is assigned to SiO4, and the fitting peaks centered at 103.5 and 102.4 eV are ascribed to silicon oxycarbide species of SiCO3 and SiC2O2.39,40 The atomic percentage of SiCO3, SiC2O2 and SiO4 is 55.90%, 12.68% and 31.42%, respectively. The above analyses convincingly reveal that Si–O–C microspheres are composed of carbonaceous matrix and silicon oxycarbide units, which is also matching well with FTIR results (Fig. 2b).
The morphology, microstructure and element distribution of microalgae carbon and Si–O–C microspheres were further investigated by scanning electron microscope (SEM), transmission electron microscope (TEM) and energy-dispersive X-ray spectra (EDS). As shown in Fig. 4a and b, microalgae carbon has the spherical morphology with a smooth surface. Meanwhile, Si–O–C microspheres have a distinct spherical shape as well, implying the morphology of microalgae is perfectly replicated after the calcination (Fig. 4c and d). However, the particle size of Si–O–C microspheres is much larger than that of microalgae carbon. The distributions of the average diameters of microalgae carbon and Si–O–C microspheres also are calculated by a statistical treatment from SEM images. As shown in Fig. S1,† the average diameter of microalgae carbon is about 15 μm. In contrast, the average diameter of Si–O–C microspheres is greatly increased, which mainly concentrated in 25 μm. This drastic change may be resulting from supercritical CO2 fluid served as superior solvent. Because supercritical CO2 fluid has excellent pressure-tunable dissolving power and super penetration ability, it can not only efficiently carry Si into microalgaes, but also intercalate into the interlayers and expand the interlayers.
 | ||
| Fig. 4 (a) and (b) SEM images of microalgae carbon. (c) and (d) Si–O–C microspheres. | ||
Fig. 5 display the cross-section SEM images of microalgae carbon and Si–O–C microspheres. As shown in Fig. 5, both microalgae carbon and Si–O–C microspheres exhibit the typical hollow structure. This phenomenon could be resulting from the pyrolysis of the tender core of microalgaes during the calcination.31 Furthermore, EDS dot-scanning results also are provided in Fig. 5c and d. The main elements in microalgae carbon are C and O. It still can be detected trace amounts of Si that is mainly arising from the cell wall of microalgae. In contrast, C, O and Si are the main elements in Si–O–C microspheres. And the atomic ratio is similar to XPS results. Additionally, the atomic ratio of C, O and Si at the different spots is very close. This result also indicates that the chemical composition of the inner side and outer side of Si–O–C microspheres is homogeneous, implying TEOS is not simply coating on the surface of microalgaes to further form a Si-rich layer.
 | ||
| Fig. 5 (a) and (b) Cross-section SEM images of microalgae carbon and Si–O–C microspheres. (c) and (d) The corresponding EDS results of microalgae carbon and Si–O–C microspheres. | ||
As shown in Fig. S2,† HRTEM images clearly demonstrate that no distinct lattice fringes can be detected in microalgae carbon and Si–O–C microspheres samples. Additionally, the corresponding selected area electron diffraction (SAED) patterns (the insets of Fig. S2†) also show that there are no sharp diffraction spots or diffraction rings in microalgae carbon and Si–O–C microspheres samples. All the above results indicate that both microalgae carbon and Si–O–C microspheres have the amorphous structure, which is in good accordance with XRD results. Furthermore, the scanning transmission electron microscope (STEM) images (Fig. 6a and f) vividly depict the particle size changes of microalgae carbon and Si–O–C microspheres. Apparently, Si–O–C microspheres exhibit a noticeable volume expansion as compared with microalgae carbon, which is consisting well the SEM results (Fig. 4 and S1†). To verify the chemical composition of microalgae carbon and Si–O–C microspheres, the element distributions of Si, O and C in microalgae carbon and Si–O–C microspheres are supplied. Taken from red square area in Fig. 6a, the EDS mappings (Fig. 6c–e) of microalgae carbon demonstrate that the main elements are C and O which are dispersed homogeneously. However, the Si signal is very weak that also verifies the content of Si is extremely low in microalgae carbon. On the contrary, Si, O and C are the main elements in Si–O–C microspheres as depicted in Fig. 6h–j, confirming the formation of Si–O–C microspheres. These results also match well with the dot-scanning EDS results in Fig. 5.
 | ||
| Fig. 6 (a) STEM image of microalgae carbon. (b)–(e) EDS elemental mapping results of carbon, oxygen and silicon taken from region 1 in (a). (f) STEM image of Si–O–C microspheres. (g)–(j) EDS elemental mapping results of carbon, oxygen and silicon taken from region 2 in (f). | ||
On the basis of the above morphology, microstructure and chemical composition analyses, a schematic diagram is provided to illustrate the formation mechanism of Si–O–C microspheres as illustrated in Fig. 7. On the one hand, supercritical CO2 fluid is a promising green solvent with extremely high pressure-tunable dissolving power. So supercritical CO2 fluid could easily dissolve TEOS at the molecular level, and it will guarantee the ability to realize the efficient mass transfer. On the other hand, the viscosity and diffusivity of supercritical CO2 fluid are similar to gas while its density is closer to liquid. Supercritical CO2 fluid can penetrate into the interlayers or nano-gaps/pores of microalgaes with high efficiency. So CO2 molecules and TEOS molecules will synchronously be intercalated into the interlayers and nano-gaps/pores of microalgaes. And this process will evoke a rapid expansion as well. When exposed to an abrupt decrease in pressure, the intercalated CO2 molecules decompose into CO2 gas immediately, resulting in a large pressure difference between the interlayers and nano-gaps/pores of microalgaes and ambient environment. Meanwhile, TEOS will remain at the interlayers and nano-gaps/pores of microalgaes, achieving the uniform dispersion of TEOS in microalgaes. Subsequently, the obtained TEOS/microalgaes precursors will be successfully converted into Si–O–C microspheres during the calcination process. Thus an expansion–impregnation–conversion mechanism can be proposed.
 | ||
| Fig. 7 Schematic diagram illustrating the expansion–impregnation–conversion mechanism of synthesizing Si–O–C microspheres. | ||
The electrochemical performance of microalgae carbon and Si–O–C microspheres is evaluated with CR2025 coin-typed cells. Fig. 8a reveals the galvanostatic charge–discharge profiles of Si–O–C microspheres at a current density of 0.1 A g−1 between 0.01 and 3.0 V. The first discharge and charge capacities of Si–O–C microspheres are 954.7 and 605.3 mA h g−1, respectively. The irreversible capacity loss is ∼352 mA h g−1 with a low coulombic efficiency of 63.4%. Meanwhile, the discharge and charge capacities of the second cycle are 505.7 and 500.6 mA h g−1. The coulombic efficiency is drastically increased to 99%. Moreover, a long sloping plateau below 0.4 V can be seen during the first discharge process. However, the discharge profiles from the second cycle onward become more sloping, and no visible plateau can be seen, indicating the occurrence of irreversible reactions during the first cycle. The irreversible discharge plateau and low coulombic efficiency in the first cycle are mainly attributed to the existence of SiO-rich sites in Si–O–C microspheres. According to previous literatures, these SiO-rich sites are energetically unfavorable which will cause the irreversible capacity.5,8
 | ||
| Fig. 8 (a) Charge–discharge profiles of Si–O–C microspheres at a low current density of 0.1 A g−1 in the potential range from 0.01 to 3.0 V. (b) CV curves of Si–O–C microspheres at a scan rate of 0.1 mV s−1. (c) Charge–discharge profiles of Si–O–C microspheres at various current densities. (d) The rate performance of Si–O–C microspheres and microalgae carbon. (e) Long-term cycling performance of Si–O–C microspheres and microalgae carbon at a current density of 0.1 A g−1. | ||
Fig. 8b shows the CV curves of Si–O–C microspheres in the potential range between 0 and 3.0 V at a scan rate of 0.1 mV s−1. No distinct anodic and cathodic peaks in the CV curves of Si–O–C microspheres. There are only two broad pseudo peaks at 0.5 V and 0.7 V in cathodic scanning and anodic scanning, respectively, which is similar to previous works.38,41 These results indicate that the lithium storage mechanism in Si–O–C system is quite different from pure Si and graphite. In fact, the lithium storage mechanism in Si–O–C materials is similar to hard carbon anodes that is correlated to the intercalation and de-intercalation processes.38,42,43 According to the nanodomains model, Si–O–C consists of three constituents: SiO4 forms the heart of the domain, the surrounding monolayer of mixed bonds of SiCnO4−n, and the graphene cage-like network that encases the domains.6 The reversible Li storage sites of Si–O–C materials mainly arise from the edges and interstitial spaces of graphene layers, directly or indirectly amorphous Si–O–C phases, micro-/nano-pores and interfacial and defect sites.5,9–11 Obviously, Si–O–C materials can store more Li compared to the conventional graphitic carbon materials. Meanwhile, all the CV curves overlap well and exhibit good reproducibility and similar accompanying with increasing cycling number, implying this amorphous Si–O–C network can mainly improve the cycling stability of the electrode.
The rate performance of Si–O–C microspheres is further investigated. As shown in Fig. 8c, the discharge–charge profiles present similar shapes at various current densities. No obvious reaction plateaus can be detected, which further support the non-alloying/de-alloying reaction mechanism in Si–O–C materials. Additionally, the rate capability and cycling stability of Si–O–C microspheres and microalgae carbon are examined by successively increasing current density from 0.1 A g−1 to 1 A g−1. As shown in Fig. 8d, the discharge and charge capacities of two samples remain stable and decrease regularly along with increasing current density. The reversible capacities of Si–O–C microspheres at 0.1, 0.2, 0.4, 0.8 and 1 A g−1 after each 10 cycles are 420.2, 338.4, 277.1, 227.6 and 211.2 mA h g−1, respectively. In contrast, microalgae carbon only can deliver 248.9, 175.7, 126.7, 78.9 and 65.3 mA h g−1 at the same current density. It is noteworthy that Si–O–C microspheres could not deliver a reversible capacity of 420.2 mA h g−1 at a low current density of 0.1 A g−1, but also retain a high capacity of 248.9 mA h g−1 at a high current density of 1 A g−1, which is even 3 times higher than that of microalgae carbon. Interestingly, Si–O–C microspheres can restore a satisfactory reversible capacity of 441.2 mA h g−1 after another 50 cycles upon reducing the current density to 0.1 A g−1. The above results clearly demonstrate Si–O–C microspheres possess the better rate performance in terms of reversible capacity and cycling stability in comparison with microalgae carbon. Furthermore, the long-term cycling performance comparison of Si–O–C microspheres and microalgae carbon is displayed in Fig. 8e. After 200 cycles at 0.1 A g−1, the discharge capacity of Si–O–C microspheres remain at 453.5 mA h g−1, whereas the coulombic efficiency steadily reaches 100%. On the contrary, the microalgae carbon shows the initial discharge capacity of 737 mA h g−1 with a low coulombic efficiency of 43.6%. The reversible discharge capacity is 292 mA h g−1 at the 100th cycle, which is only 64.8% of the reversible capacity of Si–O–C microspheres. Interestingly, the specific capacity and cycling stability of Si–O–C microspheres are also comparable to other previous literatures as shown in Fig. S3.† These enhanced reversible capacity and excellent cycling stability of Si–O–C microspheres can be attributed to two aspects. On the one hand, Si–O–C microspheres will provide more active sites for the Li storage in silicon oxycarbides so that can largely contribute to the reversible capacity. The intermediate mixed silicon oxycarbide species are also known to be electrochemically reversible with lithium and can contribute to the reversible capacity.9 On the other hand, according to the nanodomain model,6,10 the intermediate mixed silicon oxycarbide species are well enwrapped by carbonaceous matrix which could effectively alleviate the strain and stress of volume change of Li+ intercalation/de-intercalation processes. Consequently, the cycling stability and structural integrity of Si–O–C microspheres can be greatly enhanced.
Nyquist plots are recorded to investigate the electrochemical dynamic behavior of Si–O–C microspheres and microalgae carbon. As shown in Fig. 9, the EIS spectra of two samples exhibit similar profiles, which consist of a small intercept in high frequency, a depressed semicircle in high to medium frequency and a slope line in low frequency. Generally, the straight line in the low-frequency is attributed to the diffusion of the lithium ions into the active anode materials, which is the typical diffusion-controlled Warburg behavior (Zw). The semicircle in the high-frequency region represents charge-transfer resistance. The value of the diameter of the semicircle on Z′ axis implies an approximate indication of the charge transfer resistance (Rct), and the small intercept of the high-frequency semicircle on the Z′ axis can be assigned to the internal electrolyte resistance (Re).31 As indicated in Fig. 9, Si–O–C microspheres and microalgae carbon both have small Re values that are 1.92 Ω cm−2 and 1.64 Ω cm−2, implying the electrolyte has fully penetrated into electrode materials leading to an extremely low internal resistance of the battery. Furthermore, the Rct values of Si–O–C microspheres and microalgae carbon are 78.68 Ω cm−2 and 70.76 Ω cm−2, respectively. The small Rct value means the faster charge transfer in the electrochemical reactions, which will be favorable to enhance the electrode reaction kinetics and achieve high rate performance. These results also further support the good rate performance of Si–O–C microspheres.
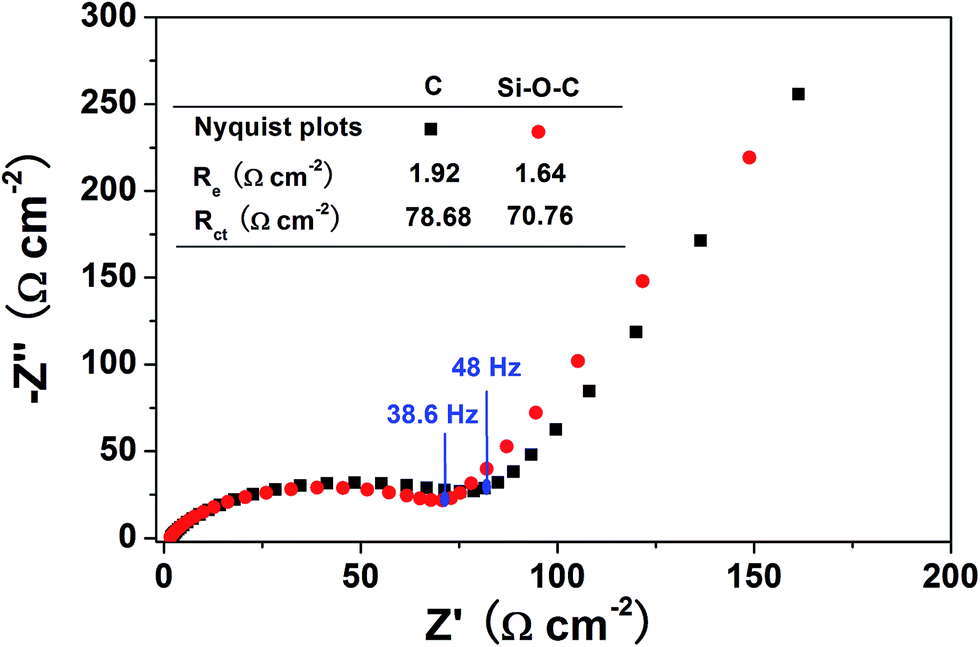 | ||
| Fig. 9 Nyquist plots of microalgae carbon and Si–O–C composites. | ||
4. Conclusions
In summary, we have successfully developed a feasible strategy to synthesize Si–O–C microspheres derived from microalgaes as both biological templates and carbon source with the assistance of supercritical CO2 fluid. According to microstructure characterization analyses, a novel expansion–impregnation–conversion mechanism is proposed. The obtained Si–O–C microspheres present amorphous structure in which the intermediate SiCnO4−n species are tightly embedded in amorphous carbonaceous matrix. This unique microstructure could not only provide more active sites for the Li storage in silicon oxycarbides to improve reversible capacity, but also effectively alleviate the volume expansion during the charge–discharge processes. As a result, Si–O–C microspheres exhibit high specific capacity, excellent cycling stability and good rate capability compared to the pristine microalgae carbon electrode. We believe that the successful implementation of biotemplating method with supercritical fluid technology will not only extend the synthesis methods of Si–O–C materials, but also provide the rational design and controllable synthesis to other functional materials for various applications, i.e. energy, catalysis and gas sensing.Author contributions
Y. Xia, X. Y. Tao and W. K. Zhang conceived the idea and designed this study. R. J. Yan cultivated microalgaes. R. Y. Fang and L. Y. Ruan synthesized materials and carried out electrochemical measurements. Z. Xiao, H. Huang and Y. P. Gan performed material characterizations. C. Liang designed and fabricated the supercritical CO2 fluid reaction device. Y. Xia and J. Zhang prepared figures. Y. Xia and R. Y. Fang co-wrote the manuscript. All authors discussed the results and commented on the manuscript.Acknowledgements
The authors thank financial support from the National Natural Science Foundation of China (51572240 and 21403196), Natural Science Foundation of Zhejiang Province (LQ14E020005, LY14E090008, LY15B030003 and LY16E070004), Science and Technology Department of Zhejiang Province (2016C31012) and Scientific Research Foundation of Zhejiang Provincial Education Department (Y201432424).References
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- J. B. Goodenough and K. S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 CAS.
- M. Winter, J. O. Besenhard, M. E. Spahr and P. Novμk, Adv. Mater., 1998, 10, 725–763 CrossRef CAS.
- M. Wilamowska, V. S. Pradeepb, M. Graczyk-Zajac, R. Riedel and G. D. Sorarùb, Solid State Ionics, 2014, 260, 94–100 CrossRef CAS.
- A. Saha and R. Raj, J. Am. Ceram. Soc., 2006, 89, 2188–2195 CAS.
- A. M. Wilson, G. Zank, K. Eguchi, W. Xing, B. Yates and J. R. Dahn, Chem. Mater., 1997, 9, 2139–2144 CrossRef CAS.
- A. M. Wilson, G. Zank, K. Eguchi, W. Xing and J. R. Dahn, J. Power Sources, 1997, 68, 195–200 CrossRef CAS.
- X. Liu, M. C. Zheng and K. Xie, J. Power Sources, 2011, 196, 10667–10672 CrossRef CAS.
- J. Kaspar, M. Graczyk-Zajac and R. Riedel, J. Power Sources, 2013, 244, 450–455 CrossRef CAS.
- V. S. Pradeepa, M. Graczyk-Zajacb, R. Riedelb and G. D. Soraru, Electrochim. Acta, 2014, 119, 78–85 CrossRef.
- J. Kaspar, M. Graczyk-Zajac, S. Lauterbach, H. J. Kleebe and R. Riedel, J. Power Sources, 2014, 269, 164–172 CrossRef CAS.
- H. Fukui, Y. Harimoto, M. Akasaka and K. Eguchi, ACS Appl. Mater. Interfaces, 2014, 6, 12827–12836 CAS.
- V. S. Pradeep, M. Graczyk-Zajac, M. Wilamowska, R. Riedel and G. D. Soraru, Solid State Ionics, 2014, 262, 22–24 CrossRef CAS.
- J. K. Li, K. Lu, T. S. Lin and F. Y. Shen, J. Am. Ceram. Soc., 2015, 98, 1753–1761 CrossRef CAS.
- X. Liu, K. Xie, C. M. Zheng, J. Wang and Z. Q. Jing, J. Power Sources, 2012, 214, 119–123 CrossRef CAS.
- X. Liu, K. Xie, J. Wang, C. M. Zheng and Y. Pan, J. Mater. Chem. A, 2012, 22, 19621–19624 RSC.
- H. Nara, T. Yokoshima, M. Otaki, T. Momma and T. Osaka, Electrochim. Acta, 2013, 110, 403–410 CrossRef CAS.
- T. Hang, D. Mukoyama, H. Nara, T. Yokoshima, T. Mommaa, M. Li and T. Osaka, J. Power Sources, 2014, 256, 226–232 CrossRef CAS.
- X. Y. Tao, Y. P. Li, J. Du, Y. Xia, Y. C. Yang, H. Huang, Y. P. Gan, W. K. Zhang and X. D. Li, J. Mater. Chem., 2011, 21, 9095–9102 RSC.
- J. Du, Y. C. Yang, Z. Fan, Y. Xia, X. J. Cheng, Y. P. Gan, H. Hang, L. X. Dong, X. D. Li, W. K. Zhang and X. Y. Tao, J. Alloys Compd., 2013, 560, 142–146 CrossRef CAS.
- Q. Zhen, H. Huang, J. Du, T. Feng, W. K. Zhang, Y. P. Gan and X. Y. Tao, J. Phys. Chem. C, 2013, 117, 13770–13775 Search PubMed.
- J. Du, Q. Q. Li, Y. Xia, X. J. Cheng, Y. P. Gan, H. Huang, W. K. Zhang and X. Y. Tao, J. Alloys Compd., 2013, 581, 128–132 CrossRef CAS.
- X. Y. Tao, L. X. Dong, X. N. Wang, W. K. Zhang, B. J. Nelson and X. D. Li, Adv. Mater., 2010, 22, 2055–2059 CrossRef CAS PubMed.
- X. Y. Tao, J. Du, Y. C. Yang, Y. P. Li, Y. Xia, Y. P. Gan, H. Huang, W. K. Zhang and X. D. Li, Cryst. Growth Des., 2011, 11, 4422–4426 CAS.
- Z. Qiu, H. Huang, J. Du, X. Y. Tao, Y. Xia, T. Feng, Y. P. Gan and W. K. Zhang, J. Mater. Chem. A, 2014, 2, 8003–8008 CAS.
- X. Y. Tao, J. T. Zhang, Y. Xia, H. Huang, J. Du, H. Xiao, W. K. Zhang and Y. P. Gan, J. Mater. Chem. A, 2014, 2, 2290–2296 CAS.
- X. Y. Tao, W. C. Chai, F. Q. Xu, J. M. Luo, H. Xiao, C. Liang, Y. P. Gan, H. Huang, Y. Xia and W. K. Zhang, Electrochim. Acta, 2015, 169, 159–167 CrossRef CAS.
- W. J. Zhu, H. Huang, W. K. Zhang, X. Y. Tao, Y. P. Gan, Y. Xia, H. Yang and X. Z. Guo, Electrochim. Acta, 2015, 152, 286–293 CrossRef CAS.
- Y. Xia, W. K. Zhang, Z. Xiao, H. Huang, H. J. Zeng, X. R. Chen, F. Chen, Y. P. Gan and X. Y. Tao, J. Mater. Chem., 2012, 22, 9209–9215 RSC.
- Y. Xia, Z. Xiao, X. Dou, H. Huang, X. H. Lu, R. J. Yan, Y. P. Gan, W. J. Zhu, J. P. Tu, W. K. Zhang and X. Y. Tao, ACS Nano, 2013, 7, 7083–7092 CrossRef CAS PubMed.
- Y. Xia, W. K. Zhang, H. Huang, Y. P. Gan, Z. Xiao, L. C. Qian and X. Y. Tao, J. Mater. Chem., 2011, 21, 6498–6501 RSC.
- X. Y. Tao, R. Wu, Y. Xia, H. Huang, W. C. Chai, T. Feng, Y. P. Gan and W. K. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 3696–3702 CAS.
- C. Aymonier, A. Loppinet-Serani, H. Reveron, Y. Garrabos and F. Cansell, J. Supercrit. Fluids, 2006, 38, 242–251 CrossRef CAS.
- M. M. Rebolloso-Fuentes, A. Navarro-Perez, F. Garcia-Camacho, J. J. Ramos-Miras and J. L. Guil-Guerrero, J. Agric. Food Chem., 2001, 49, 2966–2972 CrossRef CAS PubMed.
- J. Kaspar, C. Terzioglu, E. Ionescu, M. Graczyk-Zajac, S. Hapis, H. J. Kleebe and R. Riedel, Adv. Funct. Mater., 2014, 24, 4097–4104 CrossRef CAS.
- G. Liu, J. Kaspar, L. M. Reinold, M. Graczyk-Zajac and R. Riedel, Electrochim. Acta, 2013, 106, 101–108 CrossRef CAS.
- M. Halim, C. Hudaya, A. Y. Kimad and J. K. Lee, J. Mater. Chem. A, 2016, 4, 2651–2656 CAS.
- G. D. Sorarh, G. D'Andrea and A. Glisenti, Mater. Lett., 1996, 27, 1–5 CrossRef.
- F. Ji, Y. L. Li, J. M. Feng, D. Su, Y. Y. Wen, Y. Feng and F. Hou, J. Mater. Chem., 2009, 19, 9063–9067 RSC.
- M. Graczyk-Zajac, L. Toma, C. Fasel and R. Riedel, Solid State Ionics, 2012, 225, 522–526 CrossRef CAS.
- H. Fukui, H. Ohsuka, T. Hino and K. Kanamura, ACS Appl. Mater. Interfaces, 2010, 2, 998–1008 CAS.
- H. Fukui, H. Ohsuka, T. Hino and K. Kanamura, J. Electrochem. Soc., 2013, 160, A1276–A1281 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Particle size distributions, HRTEM images of microalgae carbon and Si–O–C microspheres, electrochemical performance comparison of various Si–O–C materials. See DOI: 10.1039/c6ra13560a |
| This journal is © The Royal Society of Chemistry 2016 |